Court records show the FDA failed at reviewing submitted PMTA data as required and only looked for specific studies.
By Timothy S. Donahue
The term “fatal flaw” was used by the U.S. Food and Drug Administration for premarket tobacco product application (PMTA) submissions that didn’t have specific studies. The term has been at the center of nearly all lawsuits filed against the FDA for its handling of the PMTA process.
In court records reviewed by Voice Voice submitted in the Triton Distribution v. U.S. FDA case requesting a stay of the marketing denial order (MDO) the e-liquid manufacturer received from the FDA, the regulatory agency submitted an administrative record for the review of Triton’s PMTA that shows the agency did not fully review all PMTA data submitted, as required by law, but instead only looked for specific studies relating to flavors and youth use.
A memo dated July 9, 2021, written by Anne Radway, the associate director of the FDA’s Center for Tobacco Products’ Office of Science, states that “based on the information available to date, FDA has determined this evaluation requires evidence that can demonstrate whether an applicant’s new non-tobacco flavored product(s) will provide an incremental benefit to adult smokers relative to the applicant’s tobacco-flavored product(s). In particular, the evidence necessary for this evaluation would be provided by either a randomized controlled trial (RCT) or a longitudinal cohort study. The absence of these types of studies is considered a fatal flaw, meaning any application lacking this evidence will likely receive a marketing denial order.”
Radway goes onto explain that due to the large number of PMTAs received, the agency would only conduct a Fatal Flaw review of PMTAs for non-tobacco flavored ENDS products.
“The Fatal Flaw review is a simple review in which the reviewer examines the submission to identify whether or not it contains the necessary type of studies. The Fatal Flaw review will be limited to determining presence or absence of such studies; it will not evaluate the merits of the studies,” Radway states. “To decrease the number of PMTAs without final action by September 9, 2021, [Office of Science] used a database query to identify the top twelve manufacturers with the largest number of pending PMTAs [in the substantive review stage of the process] … Following completion of filing those applications that are filed will immediately initiate Fatal Flaw review.”
Radway also states that for the remaining PMTAs not in [substantive review] for non-tobacco flavored e-liquid products, FDA will send a “General Correspondence letter requesting the applicant to confirm if their PMTA contains such evidence and, if so, to direct FDA to the location in the application where the studies can be found.”
During the first day of TMA’s “From Chance to Change” webinar on Nov. 17, panelists were disturbed by the findings that the agency, rather than reviewing a submission on its merits, simply searched for the presence or absence of certain studies.
Brittany Cushman
Brittani Cushman, senior vice president, general counsel and secretary at Turning Point Brands said that the “idea that so many of the applications were reviewed with an eye toward this so-called fatal flaw analysis” didn’t “feel like the right direction” for the PMTA review process.
The FDA admitted it made an error in TPB’s PMTA review and TPB did in fact submit studies that the agency decided during the PMTA process were needed, after saying for years the studies were not required. The FDA then rescinded TPB’s MDO and placed its applications back into substantive review. The agency has since rescinded or a court has stayed MDO’s for 10 companies and the agency is currently facing at least 45 lawsuits for it handling of the PMTA process. This is in addition to the dozens of requests for supervisory review.
“The way the review process has played out this far, really, feels like the incentive structure in the nicotine industry has been placed on its head,” explained Cushman. “It seems that the lower-risk products are receiving heightened scrutiny, kind of an opaque direction as to what’s sufficient. And it just doesn’t feel like these products are getting a kind of equitable treatment in the space.“
Triton Distribution had their MDO stayed by the 5th Circuit Court of Appeals with the court holding that Triton is likely to succeed on the merits of its case because the FDA “changed its regulatory requirements” and that this “switcheroo” to now require a randomized controlled trial and/or a longitudinal cohort study – which the Agency previously stated on numerous occasions would not be required – was arbitrary and capricious under the Administrative Procedure Act.
The court stated that the FDA failed to “reasonably consider the relevant issues and reasonably explain” the MDO. The Court further noted that FDA failed to consider Triton’s marketing plan, surveys, and evidence of potential benefits of flavored e-cigarettes. FDA also “failed to consider the company’s legitimate reliance interests, as Triton relied on FDA’s statements made in numerous public meetings, guidance documents and rulemakings” that it did not expect applicants would need to conduct long-term studies to support their PMTAs.
Cushman told webinar watchers that, at the end of the day, the FDA’s regulatory treatment of the various product categories is to the detriment of the adult smoker.
“We’re all down in the weeds of this. But it’s difficult to see how we ended up at this point. And it certainly can’t be where anyone wanted this process to play out,” she said. “I think this has led to a lot of detrimental outcomes. You have adults seeing a large number of vapor products being deemed as not appropriate for the protection of public health while seeing no change in [combustible] cigarette offerings in their local C-store … This is being celebrated not only by those who are ignorant to the science, but more perversely, those [who understand the science and should] know better.”
For more on this session from TMA 2021 read the next issue of Vapor Voice coming in mid-December.
Synthetic nicotine could be considered a component of e-cigarettes, which would allow for the product to be regulated by the U.S. Food and Drug Administration. Mitch Zeller, director of the FDA’s Center for Tobacco Products, said the agency was concerned about the use of synthetic nicotine to avoid regulation and enforcement and is considering its options in dealing with its use.
Mitch Zeller, director of the FDA’s Center for Tobacco Products
On Nov. 17, the first day of TMA’s “From Chance to Change” webinar, Zeller said that the agency is charged with regulating tobacco products, which according to the Tobacco Control Act is anything that’s “made or derived from tobacco that is intended for human consumption, including any component, part or accessory of a tobacco product.” Zeller said that components and parts could include everything from coils and batteries to all the ingredients comprised in producing e-liquids (such as flavorings and vegetable glycerin) even if the product does not contain nicotine.
“That’s an assessment that we need to make on a case-by-case basis based upon the totality of all the information that we have,” said Zeller, adding that another challenge is that synthetic nicotine is now of such high quality and complexity that it has become difficult to differentiate it from nicotine derived from natural tobacco. “Historically, that hasn’t been a problem,” he said. “It’s not a problem now, but it could become a challenge for us going forward.”
Zeller explained that nicotine is comprised of two isomers: R and S. Tobacco-derived nicotine is 99 percent S, and early synthetic nicotine had a 50-50 split between R isomers and S isomers. However, newer versions of synthetic nicotine have much higher proportions of S isomers (as high as 99.9 percent pure), making it harder to tell synthetic apart from natural nicotine. Tobacco-derived nicotine is also becoming higher in quality.
“Tobacco-derived nicotine is now being made available at a higher quality … pharmaceutical grade from a purity standpoint. And with that, it may be harder for us to see that chemical fingerprint, if you will, whether it’s tobacco DNA or tobacco-specific nitrosamines,” he said. “We could see this as a problem going forward. Coupled with the clear intent of certain companies to do this to evade FDA regulation … We are concerned about what this means for product regulation, for the public health, and a product like Puff Bar proudly proclaiming its use of synthetic nicotine, [and] being the number-one brand used by youth.”
In the short term, Zeller said the FDA is talking internally about how to best address the growing number of products that are using synthetic nicotine to skirt FDA regulation. He said the agency is also responding to questions from Congress about synthetic nicotine and providing technical assistance to members when asked.
“There are a lot of companies out there that pride themselves on playing by the rules. They have every right to expect that the playing field is going to be level. That’s where we come in with our compliance and enforcement authorities,” Zeller said. “We agree that one of the most important things that we can do, using our compliance and enforcement tools, is to level the playing field and to have our actions [in the e-cigarette space], hopefully, serve as a deterrent. There’s nothing that I can say from a compliance enforcement standpoint on synthetic nicotine other than we have ongoing investigations.”
For more on Zeller’s speech at TMA read the next issue of Vapor Voice coming in mid-December.
A divided panel of the U.S. Court of Appeals for the Sixth Circuit rejected Breeze Smoke LLC’s application of a stay of the U.S. Food and Drug Administration’s order Friday, denying the company’s premarket tobacco product application (PMTA) for some of its vaping products.
In Breeze Smoke LLC v. FDA, the Sixth Circuit rejected the Fifth Circuit’s conclusion that the FDA had orchestrated a “surprise switcheroo” in the PMTA review process. This creates an interesting circuit split that might attract Supreme Court interest, according to Reason’s Jonathan Hadler.
The Sixth Circuit’s order, on behalf of Judges Moore and Gilman, concluded that the FDA had never committed itself to accepting PMTA applications for flavored vaping products that lacked long-term studies. Rather, the FDA had merely indicated that “it might accept evidence other than long-term studies, if that evidence had sufficient scientific underpinnings to meet the [Tobacco Control Act’s] statutory mandate of demonstrating that flavored ENDS devices are appropriate for the protection of public health” (emphasis in original).
Thus the court concluded that Breeze Smoke had failed to demonstrate the strong likelihood of success on the merits necessary to support a stay. Judge Kethledge dissented, noting his agreement with the Fifth Circuit’s decision in Wages and White Lion Investments LLC v USFDA.
While rejecting Breeze Smoke’s stay request, the Sixth Circuit panel did note some concern with the FDA’s handling of the company’s application, particularly its “formulaic consideration” of Breeze Smoke’s plans to prevent marketing to youth. This failing, and the impact of a PMTA denial on Breeze Smoke’s business were still not enough to convince a majority of the panel to enter a stay however.
The U.S. Food and Drug Administration has rescinded the marketing denial order (MDO) issued Sept. 15, 2021, for Humble Juice Co.’s flavored e-liquid products, the company announced on Nov. 5.
Humble had filed a petition in October with the U.S. Court of Appeals for the Ninth Circuit, challenging the FDA’s decision and seeking to have the MDO vacated. Following the receipt of the rescission letter, Humble withdrew its petition as FDA’s rescission of Humble’s MDO places the brand’s flavored e-liquids back into the PMTA review process and provides Humble with a pathway to market its products while its PMTAs are pending.
FDA’s rescission letter states that upon further review it identified information contained in Humble’s PMTA that requires additional evaluation such as “randomized controlled trials comparing tobacco-flavored ENDS to flavored ENDS as well as several cross-sectional surveys evaluating intentions to use or likelihood of use in current smokers, current ENDS users, former tobacco users, and never users.”
The agency also stated that due to the unusual circumstances, it “has no intention of initiating an enforcement action” against any of Humble’s flavored e-liquid products with pending PMTAs. Humble will continue to market its products while its application remains in the review process.
“FDA’s decision to rescind the MDO re-instills our faith in this challenging but science-based regulatory process,” said Humble CEO Daniel Clark. “We remain confident in and proud of our extensive PMTA submission. We are committed to working with the FDA to obtain marketing orders for the products submitted in our initial PMTAs in order to provide Humble’s adult consumers with flavor-filled and affordable e-juice long into the future.”
Confronted with an unexpected large volume of premarket tobacco applications, the U.S. Food and Drug Administration subjected some premarket tobacco product applications (PMTAs) to only a superficial review, according to documents obtained by Filter.
The PMTA review process comprises three phases: Phase I (Acceptance), which essentially means an application has been received; Phase II (Notification or Filing), which entails acknowledging a company had enough information for its applications to be formally filed; and Phase III (Review), which involves a substantive scientific evaluation, followed by a marketing granted order or marketing denial order (MDOs).
Overwhelmed by the large number of PMTAs and facing a court-ordered deadline of Sept. 9, 2021, the FDA in effect opted for a shortcut, according to Filter.
The publication cites a memorandum signed on July 9 by Matthew Holman, the director of FDA Center for Tobacco Products Office of Science (OS). “Considering the large number of applications that remain to be reviewed by the September 9, 2021, deadline, OS will conduct a Fatal Flaw review of PMTAs not in Phase III for non-tobacco-flavored ENDS products,” it reads.
The Fatal Flaw review is a simple review in which the reviewer examines the submission to identify whether or not it contains the necessary type of studies. “The Fatal Flaw review will be limited to determining presence or absence of such studies; it will not evaluate the merits of the studies,” the memorandum states.
Filter suggests that CTP reviewers created what’s probably a new method to get through a backlog of millions of PMTAs, searched those applications for longitudinal cohort studies and randomized clinical trials without evaluating any other evidence, and for applications lacking them, did not advance them beyond Phase II and just sent out templated MDOs.
The U.S. Food and Drug Administration devastates small businesses with a plethora of marketing denial orders.
By Timothy S. Donahue
At press time, the U.S. Food and Drug Administration had yet to approve an electronic nicotine-delivery system (ENDS) product for sale in the U.S. But it had killed much of the U.S. market for such products. As of Sept. 23, the agency had issued 323 marketing denial orders (MDOs) accounting for more than 1,167,000 flavored vaping products. In addition, the FDA previously refused to accept (RTA) or refused to file (RTF) a significant share of the nearly 7 million applications it received from more than 500 companies.
At least four lawsuits contesting MDOs have been filed in the 2nd, 4th, 6th and 11th Circuit Courts of Appeals against the FDA. Turning Point Brands (TPB) filed a petition for review with the United States Court of Appeals for the 6th Circuit. The petition forced the FDA to provide an administrative record for its decisions on PMTAs. TPB sells various flavored e-liquids marketed under the Solace, VaporFi and Vapor Shark brands.
In a surprise move as this magazine was going to press, the FDA rescinded Turning Point Brands’ MDO. The FDA admitted it made an error in TPB’s PMTA review and TPB did in fact submit studies that the agency decided during the PMTA process were needed, after saying for years the studies were not required. The FDA had not yet responded to the remaining cases as of press time.
“Upon further review of the administrative record, FDA found relevant information that was not adequately assessed,” the FDA letter to TPB states. “Specifically, your applications did contain randomized controlled trials comparing tobacco-flavored ENDS to flavored ENDS as well as several cross-sectional surveys evaluating patterns of use, likelihood of use, and perceptions in current smokers, current ENDS users, former tobacco users, and never users, which require further review.”
TPB was asking the court to review the FDA order “on the grounds that it is arbitrary and capricious, an abuse of discretion, contrary to the Federal Food, Drug and Cosmetic Act, as amended by the Family Smoking Prevention and Tobacco Control Act of 2009, and otherwise not in accordance with law.” The company requests the court “vacate or modify” the FDA order and asks that TPB be allowed to “continue to market the products subject to the challenged order.” Bidi Vapor filed a similar suit in the U.S. Court of Appeals for the 11th Circuit, BMF (Bad Modder Fogger) filed in the 4th Circuit and Magellan Technology, parent to DemandVape, has filed in the 2nd Circuit (those lawsuits are still active).
Credit: CASAA
In addition to its arbitrary claim, Magellan also claims in its court petition that the “FDA’s issuance of an MDO in the absence of a finalized rule” setting forth the required contents of a PMTA is unlawful. “FDA’s adoption of a comparative efficacy standard for the granting of a marketing order for non-tobacco- and non-menthol-flavored ENDS products versus tobacco-flavored ENDS products is, in reality, a disguised tobacco product standard that has been adopted and is being applied by FDA through adjudication rather than adopted through notice-and-comment rulemaking,” states Magellan’s petition.
According to Mitch Zeller, the director of the FDA’s Center for Tobacco Products (CTP), many of the accepted applications ultimately received an RTF letter because they did not include required information. “For example, companies received RTF letters for not including required content such as ingredient listings, labels for each product to be marketed or adequate environmental assessments,” he wrote.
In a joint news release with Zeller and acting FDA Commissioner Janet Woodcock, the FDA explained that the applications from many MDO recipients “lacked sufficient evidence that they have a benefit to adult smokers sufficient to overcome the public health threat posed by the levels of youth use” of ENDS products.
The PMTAs submitted by TPB and subsequently denied market access and the brought back under review by the FDA included an in-depth toxicological review, a clinical study and studies on patterns and likelihood of use, according to a motion to stay filed by TPB on Sept. 30. “In light of the unusual circumstances,” the FDA’s Center for Tobacco Products (CTP) Director Matt Holman stated in the letter. “FDA has no intention of initiating an enforcement action” against TPB’s products that had previously received an MDO.
Many of the current lawsuits against the FDA accuse the FDA of many of the same issues TPB’s withdrawn suit claimed. For example, TPB’s stay said the agency had moved the goalposts for data needed to receive a marketing order based on what the agency “learned” from the “review [of] PMTAs for flavored ENDS so far,” according to the stay. TPB noted that the “North Star of administrative law” is that agencies cannot induce regulated parties to rely on “agency representations about regulatory requirements” then penalize them using the previously unannounced criteria after the fact.
“But that is precisely what FDA did here,” the stay motion states. “[The] FDA reasoned that TPB failed to conduct ‘a randomized controlled trial and/or longitudinal cohort study’ or other studies performed ‘over time’ to show that TPB’s specific flavored products help adult users stop smoking more than tobacco-flavored products do. Yet FDA previously deemed these studies unnecessary.”
Tony Abboud, executive director of the Vapor Technology Association, suspects the FDA made an internal policy decision to change the PMTA standard to make it impossible after the fact for a company to comply and get a flavored ENDS application approved. “I think that that decision is being implemented application by application, which I don’t believe is fair under the law,” said Abboud. “I think that the refocusing on open system flavored e-liquids is a direct result of the public and political pressure that was placed upon the FDA by Congress, which expressly said they were trying to interfere with the regulatory process.”
What’s in a name?
Critics say the FDA has made several “sloppy” mistakes in reviewing PMTAs and issuing MDOs. Numerous companies say the agency was inconsistent in banning flavors based solely on the flavor’s name. Bidi Vapor’s parent, Kaival Brands, said that the agency banned its “Arctic” flavor, misidentifying it as a “not-menthol” flavor. TPB also says in its stay motion that the FDA is forcing TPB to pull nonflavored products from the market; however, the FDA’s order applies to “Authentic Tobacco” and “Bold Tobacco” yet not “Classic Tobacco” (which the FDA is still considering).
Credit: Cursed Senses
“Those are the same flavors with the same formulations; they just use different names across product lines. The same goes for ‘Ripe Tobacco’ (forbidden) and ‘Smooth Tobacco’ (reprieve) and for ‘Mint’ (banned) and ‘Mighty Menthol’ (allowed for now),” the stay explains. “It is anyone’s guess why some of these products must exit the market immediately yet others might pass muster if FDA actually reviews TPB’s studies.”
Since January 2021, the agency has issued at least 170 warning letters to firms that collectively have listed more than 17 million ENDS products with the FDA and that did not submit premarket tobacco product applications (PMTAs) for the products by Sept. 9, 2020. Applications for products manufactured by major companies, such as Vuse, Juul, Logic and blu, are still under review. During this time, the agency also granted substantial equivalence (SE) status (marketing approval) to over 350 combustible products from the cigar, pipe and hookah tobacco product categories.
Amanda Wheeler, president of the American Vapor Manufacturers Association (VMA) and the owner of Jvapes e-liquids (see “No Surrender,” page ?), assisted more than 230 small-sized to mid-sized e-liquid manufacturers in submitting PMTAs for more than 1.7 million products. Nearly all of those applications received either an RTA, RTF or an MDO.
Wheeler tweeted on Sept. 9 that it was a “tough day” for the industry because “lots of very good people who I respect deeply and who helped thousands of smokers quit got told by our government that their products were illegal. To all of you, I am so very sorry. To your customers, I am even more sorry. Our government is wrong on this.”
Before the announcement, many industry experts said that banning most e-cigarettes from the market could harm public health. In a commentary published on the Reason Foundation’s website, Guy Bentley, the organization’s director of consumer freedom research, states that the sooner that U.S. public health officials embrace vaping’s potential to improve public health by reducing smoking and smoking-related deaths, “the better off we’ll all be.” The result of shutting down a vast portion of the vape industry, he warns, will be more smoking.
Anti-vaping activists, by contrast, argued for a ban on e-cigarettes. In a recent blog post, Laurie Rubiner, executive vice president of domestic programs at the Campaign for Tobacco-Free Kids, and Linda Mendonca, president of the National Association of School Nurses and an assistant professor at the Rhode Island College School of Nursing, wrote that the “evidence is clear” that as long as any flavored e-cigarettes remain on the market, kids will get their hands on them (no reference to evidence was provided).
“To truly protect kids and end the youth e-cigarette epidemic, the FDA must eliminate the flavored and high-nicotine products—including the popular menthol flavor—that have driven this crisis,” the pair write. “Parents, educators and health advocates are counting on the FDA to take them off the shelves.”
Tom Miller, attorney general for the state of Iowa, said the FDA actions against flavors endanger public health. He said that the best science available indicates that most youths are not getting e-cigarettes from vape shops and that a significant number of adults are using products from vape shops to move away from combustible cigarettes.
“Let’s not forget the overwhelming risk to public health: The CDC [U.S. Centers for Disease Control and Prevention] estimates the burden of tobacco use in the United States is 480,000 lives a year, all of which is due to the use of cigarettes,” Miller said in a statement. “We believe in the strong, science-based regulation of alternative tobacco products, and the FDA is the best agency to undertake that task. Policymakers must strike the right balance between making accessible potentially lifesaving lower risk nicotine products while discouraging use by those who wouldn’t smoke, especially youth.”
Impacts of regulation
Several studies have suggested that if vape product sales were restricted to tobacco flavors, many would return to combustible tobacco. One study found that approximately one-third of U.S. vapers aged 18 to 34 say flavor bans would push them back to smoking traditional cigarettes. The study published in Nicotine & Tobacco Research analyzed data from February to May 2020 and looked at 2,159 young adults in Atlanta, Boston, Minneapolis, Oklahoma City, San Diego and Seattle, examining support for e-cigarette sales restrictions and the perceived impact of flavor and vaping bans.
Credit: JHVEPhoto
Two other recent studies showed similar results. A study in JAMA Pediatrics showed that following San Francisco’s flavor ban, teens were more likely to smoke than those in other school districts. A different study in Nicotine & Tobacco Research shows that teens who vape would be smoking cigarettes if vapes hadn’t become available.
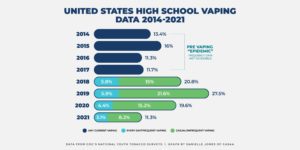
Recent evidence also seems to show that the overall youth use of e-cigarettes in the U.S. is declining. According to the 2021 National Youth Tobacco Survey (NYTS), the FDA and the CDC found that youth use of e-cigarettes fell sharply in 2021. It’s the second consecutive year of major declines. As is typical in the release of the NYTS data every year, media reports about the NYTS were all over the board. One headline read, “Big Drop in U.S. Teen Vaping with Covid Closures” while another read, “Teen Vaping Craze Shows No Sign of Slowing.”
The study shows that an estimated 11.3 percent (1.72 million) of high school students and an estimated 2.8 percent (320,000) of middle school students reported current e-cigarette use, lower than the 19.6 percent (high school) reported in 2020 and substantially lower than the 27.5 percent (high school) reported in 2019, according to previous FDA statements. Middle school vaping fell to 2.8 percent this year from 4.7 percent in 2020—a 40.4 percent decline. Middle school past 30-day vaping in 2020 fell 55.2 percent from 2019.
Chris Allen, chief scientific officer at Broughton, a contract research organization (CRO) delivering analytical, scientific and regulatory services for the ENDS industry, said that the FDA might well be using the NYTS to justify the “flurry of MDOs” issued for flavored e-liquids. He also said the majority of the companies that have fallen foul of the recent MDOs are responsible manufacturers supporting tobacco harm reduction.
“I completely accept that youth use is unacceptable; however, the issue doesn’t appear to lie primarily in open systems but a product that is currently outside the jurisdiction of FDA: a disposable containing synthetic nicotine,” Allen said. “Regardless of the product, or the source of nicotine, there’s no place for irresponsible marketing and distribution practices that keeps adding fuel to this fire. I fear that the latest action is simply going to lead to a seismic shift into the black market and unregulated (synthetic nicotine) products, which will be near on impossible for the U.S. government to control. From my personal perspective, this doesn’t seem an appropriate way to support THR [tobacco harm reduction].”
Industry representatives predict major battles at the state level. “States are just going to ban the sale of any non-FDA approved product,” said a vape shop owner, who asked not to be identified as he had not yet received an MDO. “This is just going to be a never-ending stream of court battles. I hope every company is at least considering appealing the MDO decisions. The whole PMTA process was a giant bait-and-switch.”
Manufacturers that submitted their applications by the Sept. 9, 2020, deadline but who have not yet received an MDO can effectively continue to sell their products as no ruling has been made on them; however, the FDA has made it clear that any company that does continue to sell these products will be doing so unlawfully, although they are not likely to face any enforcement action due to the agency’s limited resources.
Numerous companies are appealing their MDOs. Many are appealing MDOs they believe were wrongly issued because the PMTAs were for tobacco and/or menthol flavors. The AVM is helping its member companies file formal appeals with the FDA because the agency “in their sloppy haste, FDA not only threw out flavored products. They also threw out many [companies] [regular] tobacco and menthol flavors. We’re starting with some of those appeals specifically for what we feel were sort of administrative errors with tobacco and menthol and also working on broader appeals.”
Companies can also contact the CTP’s Office of Small Business Assistance (OSBA) with general questions regarding statutory and regulatory requirements, including the appeals process. Another option is the FDA Office of the Ombudsman, the agency’s “focal point for addressing complaints and assisting in resolving disputes between companies.”
Deanna Clark with the Clark-Esposito Law Firm stated in a blog post that each company must submit its own submission appealing the FDA decision. Companies should not send in an appeal combined with other companies, she cautioned. “Next, you want to address arguments refuting [the] FDA’s basis for your denial. It can’t just be where you’re complaining about how it’s unfair and the government sucks,” said Clark. “You need to use some rational basis behind what you’re submitting to them. And thirdly, you need to submit it to the right office and make sure it gets to the right people within the right timeframe.”
The e-cigarette saga with the FDA is far from over. Between lawsuits and appeals, many decisions may eventually be left out of the hands of the FDA entirely. The FDA’s ombudsman and appeals court judges could now decide the fate of flavored e-liquids. Congress could possibly step in and change the statutes, but many have said that is unlikely. The industry is also still waiting for decisions on the PMTAs filed by the major tobacco companies, and if anyone is approved, it may open the door for standard equivalency products. The only thing that hasn’t changed in the vaping industry is its uncertain future.
In the wake of marketing denial orders, many U.S. e-liquid manufacturers are turning to synthetic nicotine.
By Timothy S. Donahue
In 2015, Mitch Zeller, the director of the U.S. Food and Drug Administration’s Center for Tobacco Products (CTP), was asked what the FDA’s position was on synthetic nicotine. “I’ll let you figure that one out for yourselves,” he said, hinting that the agency would regulate it as a drug. Today, vaping products, especially disposable devices, using tobacco-free nicotine (TFN) are one of the fastest-growing segments on the market.
After the FDA began issuing marketing denial orders (MDOs) to companies whose premarket tobacco product applications (PMTAs) failed to satisfy the agency’s concern about youth use, many rejected applicants hinted that they would start using synthetic nicotine—nicotine made in a lab and not derived from tobacco—in their flavored e-liquids.
Vapor Salon, for example, announced on Facebook that it would be switching to synthetic nicotine less than 24 hours after the FDA ordered the company to remove its products from the market.
“The main purpose of this is to be outside of the FDA’s regulations with their hefty PMTA requirement, which takes full effect on Sept. 9, 2021, with needing an approved PMTA or your product can no longer be sold,” the company wrote.
In July 2020, Puff Bar announced that it would cease all online sales and distribution in the U.S. until further notice after receiving a warning letter from the FDA. However, the brand resumed sales on its website in February of this year with an altered product. To get around the ban on its products, Puff Bar began using tobacco-free nicotine. As of this writing, Puff Bar continues to hawk its products both on its website and in convenience stores around the U.S.
Meanwhile, the popular online vaping retail website Element Vape.com has at least 11 brands offering several synthetic nicotine e-liquids in different flavors, including fruits, cereals and candies. Pioneer e-liquid manufacturer Five Pawns reformulated its vape juice using synthetic nicotine even before the Sept. 9, 2020, PMTA submission deadline.
“Synthetic nicotine products still must abide by nationwide age restrictions, but the Center for Tobacco Products lacks the ability to regulate them as ‘tobacco products,’” said Greg Conley, president of the American Vaping Association. “Unless and until the FDA authorizes a sufficient number of flavored products to keep current ex-smokers off of cigarettes, we will support efforts by small businesses to keep offering their products to adult customers.”
Tony Abboud, executive director of the Vapor Technology Association, said that synthetic nicotine has been available and on the market since as early as 2014, and while the FDA and U.S. Congress could have elected to regulate synthetic nicotine at any time, they have chosen not to confront the issue.
“If it wasn’t for the innovation of the vapor industry, cigarette companies would not today be saying, ‘We want to get rid of combustibles.’ Synthetic nicotine is simply the next level of innovation, and it’s not surprising the government is behind it. The [U.S.] government is always behind companies in any industry that is technological and that innovates,” explains Abboud. “There’s no surprise here. There’s no loophole here. There’s no evasion here. The marketplace is what the marketplace is. It’s up to the government to figure out if and how it wants to catch up.”
Credit: Myvisuals
Anti-tobacco groups, by contrast, vowed to halt the spread of synthetic nicotine. In a recent letter to the FDA, the Campaign for Tobacco-Free Kids (CTFK), the American Academy of Pediatrics and the American Lung Association, among other organizations, argued that e-cigarette manufacturers are using “a loophole” to avoid government regulations.
“As FDA denies marketing applications for e-cigarettes, manufacturers are exploring using synthetic nicotine in order to continue marketing their products while avoiding FDA regulation,” the letter states. “This development makes it even more imperative that FDA take immediate action against illegal, synthetic nicotine products.”
Matthew Myers, president of CTFK, said synthetic nicotine is not a safer product, and his organization has sent at least three letters since 2018 to the FDA concerning synthetic nicotine products, none of which has received a response from the regulatory agency. “It is totally designed to circumvent government regulation,” he said. “The companies that have used nicotine derived from tobacco to [now] nicotine made in a laboratory are the companies whose products have been denied because of their appeal to youth and their lack of evidence that they actually help smokers quit.”
Conley said that there is a reason the CTFK’s and other letters have gone unanswered through two different presidential administrations. “Tobacco-free nicotine was invented to eliminate trace levels of impurities that are present in traditional nicotine sources, not to evade regulation. Rather than expanding the futile war on drugs to nicotine, we believe all nicotine products should be regulated as consumer products and sold only to adult consumers 21 years and over.”
Defining tobacco products
Whether the FDA will allow products with synthetic nicotine to stay on the market remains to be seen. Despite its growing popularity, the category current operates in a regulatory void. Because the product is not derived from tobacco, it does not necessarily fall under the 2009 Family Smoking Prevention and Tobacco Control Act or meet the requirements of the Federal Food, Drug and Cosmetic Act’s definition of a tobacco product.
When synthetic nicotine first appeared on the market in 2016, the product was marketed as a way to circumvent the FDA’s proposed deeming rule for next-generation tobacco products by at least one company. The FDA’s definition of “tobacco product” includes any product made or derived from tobacco [that is intended for human consumption], including any component, part or accessory of a tobacco product. E-liquids that do not contain nicotine or other substances made or derived from tobacco may still be components or parts and, therefore, subject to the FDA’s tobacco control authorities, according to the agency.
“However, it’s possible that a disposable, closed system device that contains an e-liquid with truly zero nicotine (or synthetic nicotine) would not be regulated by the FDA as a tobacco product if it is not intended or reasonably be expected to be used in such a fashion,” the FDA states on its website. “[The] FDA intends to make these determinations on a case-by-case basis, based on a totality of the circumstances.”
In late 2016, Next Generation Labs (NGL), the maker of proprietary TFN Nicotine—nontobacco derived synthetic nicotine liquid and crystals—noted court statements made by the FDA in the NicoPure Labs LLC v. U.S. Food & Drug Administration that seemed to confirm that products not made or derived from tobacco fall outside of the FDA’s deeming rule.
Credit: NDABCREATIVITY
TFN claimed that in a response brief to the court dated Nov. 1, 2016, the FDA had stated that not all nicotine-free e-liquids (NFLs) were subject to the deeming rule. “Assuming an NFL is not made or derived from tobacco, it is subject to the rule only if it meets the definition of a ‘component or part’—that is, if it is ‘intended or reasonably expected’ either … (1) to alter or affect [a] tobacco product’s performance, composition, constituents or characteristics; or (2) to be used with or for the human consumption of a tobacco product; and is not an accessory,” the FDA was quoted as having said.
Experts have also said that the FDA could potentially assert jurisdiction over synthetic nicotine as a tobacco product and argue that, when the legislation was written, nobody had the foresight to think about synthetic nicotine. Eric Lindblom, a senior scholar at Georgetown’s O’Neill Institute for National and Global Health Law and a former director of the FDA’s Center for Tobacco Products Office of Policy, said that, in response to such moves by vapor companies, the FDA could either assert jurisdiction over synthetic nicotine as a tobacco product or push for synthetic nicotine to be regulated like any other drug.
Congress could eventually pass a nationwide ban on synthetic nicotine. A more likely scenario, however, according to industry insiders, is that individual states ban the sale of synthetic nicotine products. On May 17 of this year, the governor of Alabama signed into law Act No. 2021–453, which was backed by Altria, with that purpose in mind.
The legislation, which went into effect Sept. 1, states that “no e-liquid, e-liquid in combination with an electronic nicotine-delivery system, or alternative nicotine product that, in the case of any such product, contains synthetic nicotine or nicotine derived from a source other than tobacco may be sold or otherwise distributed” in Alabama if products have not been approved by the “FDA for sale as a drug, device or combination product.”
Abboud says vapor companies may not want to face the drug regulatory pathway. “Drug protocols are absurd, and if companies cannot even survive this PMTA process, then how would they ever possibly survive the other one?” he questioned. “Are you going to blame a company that spent millions of dollars trying to comply with [the] FDA’s opaque regulatory process, find that the FDA changed the rules at the last moment after the fact, and then and you’re going to criticize that company for doing something that’s currently lawful?”
For the time being, synthetic nicotine e-liquids will likely keep flavored e-liquids on the market despite the FDA’s efforts to remove them. However, Conley warned manufacturers against publicly advertising or celebrating their decision to switch to synthetic nicotine.
“A note to manufacturers planning to use TFN—don’t make public pronouncements about what you’re planning to do over the next month. Just do it,” he tweeted. “[The] FDA may have no respect for you, but there’s no need to blast them publicly. Plenty of harm reduction advocates can handle that.”
Sidebar
What is synthetic nicotine?
A major argument for synthetic nicotine is that it is safer than tobacco-sourced nicotine. The synthetic nicotine has no tobacco-specific nitrosamines (TSNAs), the harmful, cancer-causing chemicals found in combustible tobacco products. TSNAs are formed when tobacco leaves are grown, cured, aged and processed.
Research has shown that all nicotine is highly addictive, and regardless of the form, should be consumed with caution. However, the chemical does not directly cause cancer, which instead results from inhaling the byproducts of combustion.
Whether manufactured naturally or artificially, the nicotine molecule has the same chemical structure, C10H14N2, meaning that it comprises 10 carbon atoms, 14 hydrogen atoms and two nitrogen atoms. What makes it special, independent of its origin, is that it is a “chiral” molecule: It has two stereoisomers that are mirror images of each other.
The most prevalent form is (S)-nicotine, the physiologically active variant. Its mirror isomer, (R) nicotine, also occurs in plant-derived nicotine in small amounts but is considered physiologically ineffective. Synthetic nicotine is made with a combination of niacin, ethanol, sulfuric acid and a few other chemicals.
Traditionally, a problem for the producers of synthetic nicotine has been that the production process is both complicated and expensive. As a chiral molecule, nicotine is far easier to produce as a synthetic nicotine with equal amounts of both (R) isomers and (S) isomers compared to a nearly pure (S)-nicotine.
Naturally derived nicotine and synthetic nicotine are identical on a molecular level. The differences are the individual or potential impurities. Nicotine derived from tobacco can contain potentially harmful impurities. Purification can be difficult and costly because the impurities appear structurally very similar to the nicotine molecule itself. But synthetic nicotine is virtually free of any impurities from the beginning, and none of the impurities are carcinogenic.
Currently, two types of synthetic nicotine are on the market: an (S) only synthetic nicotine and a 50 percent (S) and 50 percent (R) synthetic nicotine. In the early years of the vaping industry, the cost of the process to produce synthetic nicotine was prohibitively expensive when compared to tobacco-derived nicotine extraction methods.
Today, that’s no longer the case. Synthetic nicotine can be purchased for nearly the same price as tobacco-derived nicotine and in some instances for even less. This is due to advancements in the commercially scaled bulk production of synthetic nicotine for use in the tobacco, vaping, pharmaceutical and scientific research industries.
In November 2020, eLiquiTech, a wholly owned subsidiary of Tobacco Technology Inc. (TTI), released its recently patented SyNic synthetic (S)-nicotine. Because SyNic has greater than 99.7 percent (S), an e-liquid needs only half the amount of SyNic to create the same effect for users as current 50/50 synthetic nicotine offerings on the market.
The U.S. Food and Drug Administration has rescinded another marketing denial order (MDO), placing Fumizer’s flavored vapor products back under review, reports Filter. Fumizer received its MDO in September.
This rescission comes just weeks after the agency withdrew an MDO issued to Turning Point Brands (TPB).
In a letter to Fumizer’s, the FDA stated that “upon further review of the administrative record, FDA found relevant information that was not adequately assessed previously.”
“Specifically,” the letter states, Fumizer’s “application did contain randomized controlled trials comparing tobacco flavored ENDS [electronic nicotine-delivery systems] to flavored ENDS as well as several cross-sectional surveys evaluating patterns of use, likelihood of use and perceptions in current smokers, current ENDS users, former tobacco users and never users, which require further review.”
The FDA has indicated that it “does not intend to initiate an enforcement action” on Fumizer’s flavored vapor products returning to the market during the new review.
Many MDO recipients have complained that the FDA has been “shifting its goal posts,” during the review process, demanding certain studies that it did not appear to require before the PMTAs were filed.
According to industry insiders, the most recent MDO recission demonstrates that TPB’s successful petition for review and motion for a stay wasn’t a one-off, resulting from the legal jurisdiction it was filed in.
“A rescission in California for Fumizer is evidence of the systemic failure of the agency to ‘adequately assess’ the science and data of a wide range of small- and mid-sized applicants while giving all of their time and attention to the large companies like Juul and Reynolds,” a source told Filter
Multiple companies have challenges their MDOs. Triton, Bidi and Gripum recently received some temporary form of stay, and My Vape Order has demanded a recission due to the fact its PMTA includes some of the same data and studies that also appears in TPB’s applications.
The U.S. Food and Drug Administration has authorized the marketing of four oral tobacco products that are no longer on the market—Verve Discs Blue Mint, Verve Discs Green Mint, Verve Chews Blue Mint, and Verve Chews Green Mint, manufactured by Altria subsidiary U.S. Smokeless Tobacco Co.
“We are pleased that FDA has determined that Verve oral nicotine products are appropriate for the protection of public health,” said an Altria spokesman. “While we discontinued selling Verve on Feb. 2, 2019, we applied learnings from this successful application to our on! submissions, which remain under review by FDA. Oral nicotine products play an important role in our vision of moving beyond smoking and remain an important part of our portfolio of products to transition adult smokers away from cigarettes.”
In a press release dated Dec. 17, 2018, Altria said it would stop the production and distribution of Verve oral nicotine products based upon the expected financial performance of these products and the regulatory restrictions that burdened the company’s ability to quickly improve these products. “We do not see a path to leadership with these particular products and believe that now is the time to refocus our resources,” wrote Chairman and CEO Howard Willard at the time.
It its Oct. 19, 2021, marketing order, the FDA said it had determined the marketing of Verve products would be consistent with the statutory standard, “appropriate for the protection of the public health.” This includes a review of data showing that youth, nonsmokers and former smokers are unlikely to initiate or reinitiate tobacco use with these products.
“Ensuring new tobacco products undergo a robust premarket evaluation by the FDA is a critical part of our mission to protect the public—especially kids,” said Mitch Zeller, director of the FDA’s Center for Tobacco Products, in a statement.
“While these are mint flavored products, data submitted to the FDA show the risk for youth uptake of these particular products is low, and stringent marketing restrictions will help prevent youth exposure. Importantly, evidence shows these products could help addicted smokers who use the most harmful combusted products completely switch to a product with potentially fewer harmful chemicals.”
Wages and White Lion Investments, parent to Triton Distribution, has been granted a stay of the marketing denial order (MDO) it received from the U.S. Food and Drug Administration. The panel of judges for the U.S. Court of Appeals for the Fifth Circuit issued the order on Oct. 15 that also granted motions to expedite the appeal case and a ruling for emergency relief.
Credit: Pixelbliss
The motion granted means the company can continue to market its electronic nicotine-delivery system (ENDS) products until the court decides on the company’s appeal of the FDA’s decision to deny its premarket tobacco product applications (PMTAs).
Triton Distribution filed a motion to stay after the FDA denied the company’s PMTA, in which Triton stated that it had been irreparably harmed as a result of the FDA’s actions and faced an imminent shutdown of its business if the motion to stay had not been granted.
“Black-letter rules of administrative law prevent an agency from retroactively changing legal requirements and from doing so without accounting for reliance interests. FDA failed to satisfy these requirements when it executed an about-face on the evidence it required to support a premarket tobacco product application (“PMTA”) for a marketing order for flavored electronic nicotine delivery system (“ENDS”) products almost a year after such applications were due,” the motion states. “FDA also acted arbitrarily and capriciously by ignoring relevant evidence found in Petitioner Wages and White Lion Investments, LLC d/b/a Triton Distributions (“Triton”) PMTA and applying a double standard to its consideration of that evidence when it issued Triton a marketing denial order (“MDO”). Further, by imposing a new, across-the-board requirement that flavored ENDS products be demonstrably more effective at promoting smoking cessation than otherwise identical tobacco-flavored products, FDA acted contrary to its authority under Section 910 of the Food, Drug and Cosmetic Act (“FDCA), 21 U.S.C. § 387j, and not in accordance with law.”
At least six companies have filed lawsuits challenging the agency’s decision to make the companies remove their products from the market. Last week, the FDA rescinded the MDO issued to Turning Point Brands (TPB) and the company will be allowed to continue marketing its vapor products while the FDA re-reviews the company’s premarket tobacco product application (PMTA).
The FDA admitted it made an error in TPB’s PMTA review and TPB did in fact submit studies that the agency decided during the PMTA process were needed, after saying for years the studies were not required. “Upon further review of the administrative record, FDA found relevant information that was not adequately assessed,” reads the FDA letter to TPB. “Specifically your applications did contain randomized controlled trials comparing tobacco-flavored ENDS to flavored ENDS as well as several cross-sectional surveys evaluating patterns of use, likelihood of use, and perceptions in current smokers, current ENDS users, former tobacco users, and never users, which require further review.”