The FDA is expected to have a new leader today. Robert Califf will likely be confirmed as the next commissioner of the U.S. Food & Drug Administration (FDA) by midday Tuesday.
On Monday night, the Senate voted 49-45 to advance his nomination as part of a cloture vote, a key procedural hurdle that can also show whether or not Senate leadership has enough votes to succeed. Technically, cloture simply streamlines the vote by limiting down the time a matter can be discussed and also restricting Senators from certain actions such as amendments that are unrelated to the vote.
Robert Califf / Credit: Modern Healthcare
While this vote was close—five Republicans and one Democrat did not vote—Senate leadership typically does not call for cloture unless it believes it has the votes to pass a measure, writes Charlie Minato, an editor with Halfwheel.
He garnered the support of five Republicans, while simultaneously having five members of the Democratic caucus vote against his nomination.
The 11th Circuit Court of Appeals granted the stays to Diamond Vapor, Johnny Copper and Vapor Unlimited. The ruling was in conjunction with Bidi Vapor’s stay. The 11th Circuit handles petitions for review from vaping businesses based in Florida, Georgia and Alabama. All four companies are based in Florida.
The decision allows the companies to continue selling their tobacco harm reduction products while the lawsuits remain active. A three-judge panel heard motions from the businesses and granted the stays by a 2-1 vote. The stays don’t guarantee that the companies will succeed in their challenges to the FDA denials, but they are an encouraging sign, according to Azim Chowdhury, a partner with Keller & Heckman law firm. He said courts usually grant stays only if the plaintiff’s case has a good chance of “succeeding on its merits.”
More than 30 companies have now sued the FDA and many of those appeals will be heard in federal courts over the next few weeks. No decisions have yet to be handed down, and early decisions could affect later ones, according to several attorneys. If there are conflicting decisions in multiple courts, the FDA’s PMTA process could eventually wind up being sorted out by the Supreme Court, according to Chowdhury.
In a highly anticipated case for the vapor industry, Triton Distribution made its opening arguments Monday in its battle with the U.S. Food and Drug Administration over how the regulatory agency conducted it premarket tobacco product application (PMTA) reviews. Triton’s lawyer urged a three-judge panel of the 5th U.S. Circuit Court of Appeals in Houston to conclude the FDA could not force manufacturers to provide studies that the agency had previously stated would not be required.
“The question before the court concerns how exactly the FDA ended up denying Triton’s PMTA—with potential implications for comparable applications by many other denied companies,” said Triton’s attorney Eric Heyer, a partner at Thompson Hine.
Credit: Sergign
In August, the FDA rejected applications to market 55,000 flavored e-cigarettes, including Triton’s, and said applicants would likely need to conduct long-term studies establishing their products’ benefits to win approval, according to Reuters. The new requirement for long-term studies differed from earlier FDA guidance and was a “surprise switcheroo,” a 5th Circuit panel concluded in October when it allowed Triton to keep selling e-cigarettes until another panel could hear its appeal.
In recently released internal FDA correspondence, the agency’s scientific staff conducted “fatal flaw” reviews that only looked for the presence of the newly required long-term studies, and if those studies were not present the agency issued a marketing denial order (MDO). During oral arguments, Heyer said the FDA’s new requirement was “arbitrary and capricious, a position conservative U.S. Circuit Judge Edith Jones appeared to agree with.
“It seems to me that’s the height of arbitrariness and capriciousness, to say we are the FDA, trust us, which I might say some of us are becoming skeptical about in light of recent vaccine experiences,” she said, alluding to COVID-19 vaccines.
Heyer argued that the process the FDA established set Triton up for failure because the new requirements were only conveyed after the deadline for when PMTAs needed to be submitted (Sept.9, 2020) had passed. It was only then that the FDA indicated that applicants would likely need randomized controlled trials (RCTs) and longitudinal cohort studies to demonstrate “comparative efficacy.”
The other two judges questioned Triton’s case. U.S. Circuit Judge Gregg Costa asked whether Triton’s products, such as one called Jimmy the Juiceman Strawberry Astronaut, were really targeted to adults. “That’s supposed to be appealing to a 40-year-old?” he asked.
U.S. Circuit Judge Catharina Haynes questioned why companies like Triton did not have enough time to develop such support for their products’ health benefits for adults given the years they have had to prepare for FDA regulation. The FDA in 2016 deemed e-cigarettes to be tobacco products like traditional cigarettes subject to agency review under the Tobacco Control Act. Manufacturers were ultimately given until 2020 to seek approval to market them.
If the court disagrees with Triton’s argument, Heyer has requested that the judges “enjoin FDA from taking further adverse action on the Petitioners’ PMTAs for 18 months to allow Petitioners to conduct the necessary studies to prove comparative efficacy,” according to legal documents.
There is no timeline for a decision in the Triton lawsuit. Judges are expected to take at a minimum weeks, if not months, to make a decision.
New Jersey Congresswoman Mikie Sherrill, on Dec. 15, introduced the Clarifying Authority Over Nicotine Act of 2021 — a bipartisan bill designed to give the U.S. Food and Drug Administration (FDA) the authority to regulate synthetic nicotine products just as it regulates nicotine products made or derived from tobacco. In a press release, Rep. Sherrill stated, “This bill will ensure all tobacco products, including products made with synthetic nicotine, are regulated by the FDA in order to protect kids in our communities and those who may seek to use these products.”
Credit: DedMityay
In a blog post, Bryan Haynes and Michael Jordan, attorneys with Troutman Pepper, state that, as it stands, the Federal Food, Drug, and Cosmetic Act (FDCA) defines “tobacco product” as “any product made or derived from tobacco that is intended for human consumption, including any component, part, or accessory of a tobacco product.” 21 U.S.C. § 321(rr)(1) (emphasis added). As the FDA concedes on its website, “it’s possible that a disposable, closed system device that contains an e-liquid with truly zero nicotine (or synthetic nicotine) would not be regulated by the FDA as a tobacco product.”
That said, there are other ways FDA might try to regulate synthetic nicotine, whether under its authority to regulate a “component” of a tobacco product or as a “drug.” In November, FDA Center for Tobacco Products Director Mitch Zeller discussed the “component” aspect of the FDCA’s definition of “tobacco product” and suggested “that components and parts could include everything from coils and batteries to all the ingredients comprised in producing e-liquids (such as flavorings and vegetable glycerin) even if the product does not contain nicotine.” He added, “That’s an assessment that we need to make on a case-by-case basis based upon the totality of all the information that we have.”
FDA could also seek to regulate synthetic nicotine as a drug. The FDCA defines drug, among other things, as “articles (other than food) intended to affect the structure or any function of the body.” 21 U.S.C. § 321(g)(1). To the extent synthetic nicotine is intended to affect a consumer’s body, FDA could attempt to assert jurisdiction. Indeed, in the 1990s, FDA tried to regulate nicotine as a “drug” and cigarettes and smokeless tobacco as “drug delivery devices.” The Supreme Court in FDA v. Brown & Williamson Tobacco Corp. found FDA lacked such authority, but one of the Court’s key findings was that Congress had passed “tobacco-specific legislation [that] effectively ratified the FDA’s previous position that it lacks jurisdiction to regulate tobacco.” Today, things are different. The 2009 Family Smoking Prevention and Tobacco Control Act gave FDA the authority to regulate tobacco products. Should FDA regulate synthetic nicotine as a drug today, it could point to recent legislation from Congress giving FDA a role in this space more broadly. So far, however, FDA has not taken this approach.
With FDA having ordered more than five million tobacco-derived, e-cigarette products off the market, several manufacturers appear to have turned to synthetic nicotine to avoid FDA’s rigorous (and costly) premarket review process. In general, that process requires those who seek to market new tobacco products to demonstrate that their sale is “appropriate for the protection of public health” with the support of scientific evidence. 21 U.S.C. §387j(c)(4), (5). After receiving a denial from FDA of his premarket review applications, one vaping company owner took to Facebook to announce the company’s switch to synthetic nicotine, adding: “We never wanted to switch to [synthetic nicotine], but the FDA forced us to make that decision as we have so many adults relying on us [for alternatives to cigarettes].”
Lawmakers have taken notice. In November, a flurry of investigations and calls to regulate synthetic nicotine products reached a new high. On November 16, nine senators sent a letter to the FDA imploring the agency to regulate synthetic nicotine products. The authors expressed concerns that e-cigarette manufacturers like Puff Bar are switching to synthetic nicotine to skirt FDA oversight and pre-market review requirements to continue selling their products — including flavored products — that they assert appeal to youth. That same day, the North Carolina attorney general launched an investigation into Puff Bar for similar reasons. And, on November 8, the House Oversight Committer’s Subcommittee on Economic and Consumer Policy sent letters to e-cigarette manufacturers Puff Bar and Next Generation Labs LLC, requesting extensive records pertaining to the production and marketing of the companies’ synthetic nicotine products.
Time will tell if Congress will pass Rep. Sherrill’s Clarifying Authority Over Nicotine Act of 2021. Given the uptick in scrutiny of synthetic nicotine products, however, there is a strong chance Congress could give FDA a clear mandate to regulate synthetic nicotine in 2022.
E-liquid manufacturers and retailers are still figuring out how to survive the FDA’s erratic regulatory rules.
By Maria Verven
The vaping industry has been in a downward spiral ever since the U.S. Food and Drug Administration began issuing marketing denial orders (MDOs) for electronic nicotine-delivery system (ENDS) products. When a product with a premarket tobacco product application (PMTA) receives an MDO, it must immediately be pulled from store shelves and removed from the market.
The FDA has issued MDOs for nearly all the approximately 6.7 million PMTAs it received. At press time, the agency was still reviewing an estimated 80,000 products, according to Mitch Zeller, director of the FDA’s Center for Tobacco Products (see “From Chance to Change,” page ?). To date, only Phillip Morris International’s IQOS device and Heatsticks and R.J. Reynolds Vapor Co.’s Vuse Solo, along with two tobacco-flavored pod cartridges, have received marketing granted orders.
The FDA also rescinded or was ordered by a court to stay at least 10 MDOs. This has caused a massive amount of confusion in the industry, especially for vape shop owners and vapor distributors who are struggling to keep only legal products on their store shelves.
Complicating matters, many manufacturers have started using synthetic nicotine in their flavored vaping products and products that had otherwise received an MDO. Synthetic nicotine is in a regulatory void as it isn’t yet being regulated at the federal level, although the FDA has stated it may be considered a component of an e-cigarette, which would put synthetic nicotine under its purview.
Many ENDS business owners say that the industry is also still suffering from the 2019 e-cigarette or vaping use-associated lung injury (EVALI) crisis that was wrongly blamed on nicotine vaping products by the FDA and the U.S. Centers for Disease Control and Prevention. It took more than a year for both government entities to state publicly that the true culprits behind EVALI were illegal THC-based vaping products. Vapor business owners must also combat the never-ending amount of misinformation that is broadcast by anti-vaping groups and the mass media.
Business owners, additionally, have major concerns about the current nicotine tax in President Joe Biden’s Build Back Better Act (as of this writing, the bill was still in the Senate). The current version of the nicotine tax applies only to vaping products and nicotine pouches. The government would tax nicotine bought by manufacturers at the rate of $50.33 per 1,810 mg of nicotine—or 2.8 cents/mg if the bill passes with the tax included.
To get a better understanding of what is happening at the street level in the ENDS industry,Vapor Voice asked a group of manufacturers, retailers and industry leaders about their experience with the FDA and how the agency’s regulatory actions have impacted their businesses.
Vapor Voice: How have the FDA’s marketing denial orders affected your business?
Char Owen
Char Owen, vice president of American Vapor Manufacturer:I think the negative PR around vaping has caused sales to stagnate for most manufacturers and vapor shops. It has also increased the smoking rates for the first time in many years. It’s heartbreaking for us to watch people revert to smoking again.
Unfortunately, most of the industry has changed to synthetic. Over 95 percent of our sales are flavored e-liquid, and with others switching, there was no choice but to switch. We only manufacture open system e-liquids in many flavors, all created from nontobacco-derived nicotine. Our biggest selling products have always been fruit flavors.
We are trying to bring in new products, such as botanicals, that can help our customers with cravings but remove nicotine from the equation. For us, it has always been about harm reduction, nothing more.
Schell Hamel, president of The Vapor Bar:Sales were affected long before the MDO was received. This down spiral began with the media attacking all vapor products as killing people when they clearly knew it was illegal THC products and the entire vapor industry handcuffed to Juul’s reputation.
According to the MDO, all products made in our lab were denied. It seemed as if they used a rubber stamp to deny anything submitted, all without review. I heard them doing similarly across the industry, then approving Vuse.
Schell Hamel
Jay Oku, business development at Five Pawns:Hundreds if not thousands of customers have been adversely affected from these misguided PMTA, sale and shipping regulations.
We had been developing products to maximize harm reduction for years and were always fascinated with the cleanliness (free of nitrosamines) and the molecular merits of synthetic nicotine. We switched all of our domestic products to synthetic nicotine mid-2020. We are grateful to have maintained our sales volume through 2021.
We saw an increase in overall sales since making the switch to nontobacco-derived nicotine, yet we’ve also seen a longer sales cycle with new accounts due to the number of MDO products that companies are selling through to make room on their shelves. Despite a slight increase in gross sales, net numbers are relatively flat due to the increase in manufacturing and shipping costs in 2021.
Trent Bohl, owner of EZJ Rolling Equipment and Smokey Joes West:It’s logistically added challenges, and the horizon looks like a tough road ahead. While many Juice manufacturers have shifted gears to get into compliance, the shift toward disposables and the future ban of them will be tough for Vape as a whole.
From recent headlines, it seems the FDA doesn’t seem to play well unless you are Big Tobacco.
Do you think there’s a growing black market of products that are no longer legal?
Owen: I absolutely know there is a growing black market. A quick Twitter or Instagram search proves that. So far, those black market dealers have not been targeted by the FDA. Only registered manufacturers have been on their radar.
We need to support and grow the harm reduction industry instead of growing a black market. For harm reduction to be successful, it must be regulated and supported by our federal bodies. Without their support, we risk creating a dangerous environment for consumers. I have a great amount of respect for the U.K. in recognizing this.
Oku: Every day the black market continues to grow. The attrition of retailers, manufacturers and distributors is being caused by excessive rogue state taxation, the PACT Act that complicates accounting and reporting, and misguided government overreach that results in flavor bans.
Jay Oku
Numerous disposable manufacturers are selling mass quantities of vapor products through back doors. Some of these black market brands push immature non-lab-produced concoctions with cartoons on their labels. These regulations push people who benefit from tobacco harm reduction technology to inferior products and even worse, back to cigarettes.
Bohl: I have stores in New Mexico, and in Mexico, which outright banned vapes. The black market is huge in Mexico; any low-dollar mercado or corner OXXO seems to have them. The USA will follow suit I suspect, given the demand for vape. When one reflects on how the black market vape cannabis carts disrupted the industry and damaged lives and harmed the reputation of the industry, it’s just going to be that times 10.
What has been your experience with FDA inspections?
Owen: Personally, I had a good inspector, but it truly is the luck of the draw, and it hasn’t been the case for everyone. In my case, he was only there to find proof of manufacturing of any MDO products, and his paperwork was written to support that. My batching logs were not reviewed nor my manufacturing practices.
One member had their inspector show up at 7 p.m. on Halloween. Another member had the FDA come to her home and photograph her home office instead of her manufacturing establishment. He then took photographs of her neighbor’s home. There were instances where the inspector pressured staff when owners or managers were not present to make MDO’d products and then sent warning letters.
The American Vapor Manufacturer is usually involved in a warning letter meeting every couple of weeks. We even help nonmembers with those. It’s a very tricky process, and it’s good to have someone there who can be objective and help both the FDA and the manufacturer resolve the matter.
Trent Bohl
Bohl: I haven’t seen them from this industry perspective, but from the agricultural side and medical marijuana side, one thought comes to mind: brutal for some, not bad for others.
What ultimately will result from these MDOs?
Owen: What the FDA did was extremely arbitrary and capricious. I feel that anyone who can afford to challenge them in court will be able to prove that. My concern is for all the small businesses that cannot afford to do that.
If something doesn’t change, you will see manufacturers close and smoking rates rise. In almost all industries except vaping, small business is celebrated. This is a shame because those small shops are the ones with the hearts for harm reduction.
To lose those small businesses would be a devastating blow to the effort in moving this country to becoming smoke-free.
Oku: I am optimistic that the FDA will reconsider or rescind MDOs and revise their outdated ambiguous and debilitatingly cost-prohibitive PMTA process.
Since 2016, FDA action against the industry has resulted in warning letters and fines to those breaking the rules, yet there’s little to no enforcement. Many MDO products are being sold since there was an enormous glut of inventory in preparation for the September 2021 ruling.
There will invariably be an increase in synthetic nicotine products until those, too, are regulated out of the market.
Congress has been slipping anti-vaping bills into much larger spending packages, such as during the 2019 holiday break deep in the Omnibus Spending Bill. These bills implement devastating regulations that put our industry and the health of our customers in jeopardy.
Bohl: The goal stated by the FDA 15 years back was the end of combustibles. Vape could have helped that. I am buying a decent stock, fearing the day one more freedom is taken away in the name of safety.
The original “Vaping Vamp,” Maria Verven owns Verve Communications, a PR and marketing firm specializing in the vapor industry.
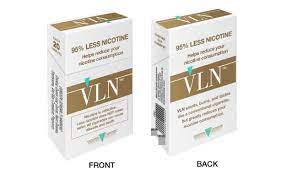
Today, the U.S. Food and Drug Administration authorized the marketing of 22nd Century Group Inc.’s “VLN King” and “VLN Menthol King” combusted, filtered cigarettes as modified risk tobacco products (MRTPs). The FDA has not yet granted a MRTP to a vaping product, even though the agency has said e-cigarettes are less harmful than combustible cigarettes. Many experts have said the low-nicotine cigarettes from 22nd Century will actually cause people to smoke more cigarettes.
These are the first combusted cigarettes to be authorized as MRTPs by the agency and the second tobacco products overall to receive “exposure modification” orders, which allows them to be marketed as having a “reduced level of, or presenting a reduced exposure to,” a substance, according to a press release.
“Our mission is to find ways to stop tobacco-related disease and death. We know that three out of four adult smokers want to quit and the data on these products show they can help addicted adult smokers transition away from highly addictive combusted cigarettes,” said Mitch Zeller, director of the FDA’s Center for Tobacco Products. “Having options like these products authorized today, which contain less nicotine and are reasonably likely to reduce nicotine dependence, may help adult smokers. If adult smokers were less addicted to combusted cigarettes, they would likely smoke less and may be exposed to fewer harmful chemicals that cause tobacco-related disease and death.”
The exposure modification orders specifically authorize the manufacturer to market “VLN King” and “VLN Menthol King” with certain reduced exposure claims regarding nicotine, including:
“95% less nicotine.” “Helps reduce your nicotine consumption.” “…Greatly reduces your nicotine consumption.”
When using any of the reduced exposure claims in the product label, labeling or advertising, the company must include, “Helps you smoke less.” The FDA also recommends that the labeling and advertising include the statement, “Nicotine is addictive. Less nicotine does NOT mean safer. All cigarettes can cause disease and death.”
Despite today’s action, these products are not considered safe or “FDA approved.” There are no safe tobacco products, so people, especially young people, who do not currently use tobacco products should not start using them or any other tobacco product, according to a press release. The exposure modification orders do not permit the company to make any other modified risk claims or any express or implied statements that convey or could mislead consumers into believing that the products are endorsed or approved by the FDA, or that the FDA deems the products to be safe for use by consumers. These orders do not allow the company to market these products with therapeutic or cessation claims.
Avail Vapor has sold the majority of its retail locations and closed its remaining stores. The company has also sold or closed its ancillary businesses. James Xu, founder of Avail, said the decision was motivated by multiple factors over several years, including what he called unclear and convoluted federal regulatory processes.
Credit: Avail
At one time, Avail Vapor was the largest family-owned vapor retailer in the U.S. with more than 100 stores in a dozen states. In January of 2020, the company split into regulatory compliance and consulting business, and a major wholesale distribution company. Those businesses have also been sold or closed.
Xu said the U.S. Food and Drug Administration’s premarket tobacco product application (PMTA) pathway was a major factor in the decision after the agency arbitrarily changed the requirements to get a PMTA approved. “It’s completely just a mess with FDA policy making and policy strategy. It just did not make any sense from day one,” Xu said. “Everything is really in this gray area. It was totally different from what our mission was and COVID is not helping any retailer.”
Xu said Avail spent more than $10 million in its bid to get regulatory approval since 2016, when the FDA set forth new compliance standards for vaping products. But the FDA rejected Avail’s applications in September and the company sued the government agency in federal appeals court. However, the FDA then stayed enforcement of the MDO on Nov. 1, pending an administrative appeal.
The economic impacts of COVID-19 also created challenges for the company, which began downsizing in August when it sold an estimated 30 of its stores to North Carolina-based competitor AMV Holdings, parent to Kure and Madvapes. It then shuttered or sold its remaining 20 stores, including five in the same city as the company’s headquarters, Richmond, Virginia, Xu said in an interview with Richmond Biz Sense.
Avail was an early entry into the vapor market, opening its first stores in 2013. By 2015, the company was producing its own e-liquids in its 37,000-square-foot office and manufacturing facility. It soon began also producing e-liquids for several other major brands. Xu said that he is not leaving the vapor industry and an announcement concerning a new project would be announced soon.
Robert Califf vowed to close the synthetic nicotine loophole if appointed commissioner of the U.S. Food and Drug Administration, according to a report by Vaping360.
During Califf’s nomination hearing on Dec. 14, Wisconsin Senator Tammy Baldwin expressed concern over reports that companies are switching to making flavored synthetic nicotine products in the wake of FDA marketing denial orders.
“As FDA commissioner, how would you work to address the rise in youth use of synthetic nicotine, and will you commit to working with Congress to ensure that the FDA has the authorities and resources it needs to crack down on these products?” Baldwin asked.
In response, Califf first noted that is crucial to appoint the right person to succeed Center for Products Director Mitch Zeller, who plans to retire in April 2022.
“Secondly,” Califf continued, “this is not limited to children. I may have some family members using synthetic nicotine, I learned as I was going through the paces here. And what people don’t realize is that there are two enantiomers of nicotine—one of which is not occurring in nature—that are in this product, and its properties are not known.
“So we’ve got to close this loophole,” Califf added, “so that we make sure that we understand the risks and benefits, and particularly deal with the issues in children.”
The Senate Health, Education, Labor & Pensions Committee will vote soon on whether to recommend Califf’s nomination to the full Senate. If the committee approves him, the former commissioner can expect full Senate confirmation to be the new commissioner soon, probably in January.
Mitch Zeller, the director of the Food and Drug Administration’s Center for Tobacco Products, plans to retire in April 2022 after serving in the post since 2013, reports The Washington Post. In a letter to staff, acting FDA Commissioner Janet Woodcock praised his work as “invaluable and instrumental” to advancing “numerous historic public health milestones in tobacco regulation.”
A graduate of Dartmouth College and the American University Washington College of Law, Zeller has been working on FDA issues for more than 30 years. He began his career as a public interest attorney in 1982 at the Center for Science in the Public Interest (CSPI). In 1988, Zeller left CSPI to become counsel to the human resources and intergovernmental relations subcommittee of the House of Representatives’ government operations committee, where he conducted oversight of enforcement of federal health and safety laws.
In 1993, Zeller joined the staff of then FDA Commissioner David Kessler. What began as a two-week assignment by Kessler to examine the practices of the tobacco industry led to his serving as associate commissioner and director of the FDA’s first Office of Tobacco Programs. Instrumental in crafting the agency’s 1996 tobacco regulations, Zeller also represented the FDA before Congress, federal and state agencies. Zeller also served as an official U.S. delegate to the World Health Organization working group for the Framework Convention on Tobacco Control.
In 2000, Zeller became executive vice president of the American Legacy Foundation. His responsibilities there included marketing, communications, strategic partnerships and, in 2002, creating the foundation’s first Office of Policy and Government Relations. That year, Zeller joined PinneyAssociates, where, as senior vice president, he provided strategic planning and communications advice.
He left PinneyAssociates in 2013 to begin his second stint at the FDA.
The U.S. Food and Drug Administration has issued warning letters to four companies marketing “wellness” vaping products. The FDA says the vaping devices contain vitamins and/or essential oils and the companies are making unproven health claims about them. Currently, no vaping products are approved by the FDA to prevent or treat any health conditions or diseases. The devices in question do not contain nicotine. The FDA calls the products “unapproved new drugs” sold in violation of sections 505(a) and 301(d) of the Federal Food, Drug, and Cosmetic Act.
Credit: FDA
“The FDA issued warning letters to companies for illegally selling these vaping products with unproven health claims. The letters provide the companies notice and request that they take prompt action to address any violations of the law,” according to an FDA press release. “If companies refuse to comply, the FDA may take enforcement actions to prevent the products from reaching consumers.”
The FDA stated that it had received complaints concerning several “wellness” products being advertised and sold to minors. “Online advertising, especially social media posts, often make false claims and cite the latest ‘scientific study,’ or do not include important details that may apply to you or allow you to make an informed decision,” the release states. “Other red flags include claims like “miracle cure” or “guaranteed results.” Remember, if a company really made a breakthrough, revolutionary health-related discovery, the news, researchers, and the government would discuss it in depth.” The letters were issued through the FDA’s Center for Drug Evaluation and Research (CDER).
The letters to Vitastick, Vitacig, NV Nutrition (NVN) and Vitamin Vape were issued on Dec. 1. Some of the unproven health or wellness claims include improving mental clarity or treating tumors or asthma. Some examples of the fraudulent product claims are:
“Fight off tumors and alleviate symptoms of chemotherapy!”
“It’s been used as a [sic] organic asthma remedy, ADHD remedy, and dementia treatment.”
“Helps prevent a type of anemia called megaloblastic anemia that makes people tired and weak.”
“Neroli oil… has long been used as a treatment against anxiety and depression, to calm the mind and soothe away tension.”
The FDA, Centers for Disease Control and Prevention, state and local health departments, and other clinical and public health partners are continuing to monitor and research vaping-associated lung injury. The FDA warns consumers to not be misled by vaping products claiming to contain “vitamins” and other “natural” ingredients or being advertised for “wellness” purposes.