Court records show the FDA failed at reviewing submitted PMTA data as required and only looked for specific studies.
By Timothy S. Donahue
The term “fatal flaw” was used by the U.S. Food and Drug Administration for premarket tobacco product application (PMTA) submissions that didn’t have specific studies. The term has been at the center of nearly all lawsuits filed against the FDA for its handling of the PMTA process.
In court records reviewed by Voice Voice submitted in the Triton Distribution v. U.S. FDA case requesting a stay of the marketing denial order (MDO) the e-liquid manufacturer received from the FDA, the regulatory agency submitted an administrative record for the review of Triton’s PMTA that shows the agency did not fully review all PMTA data submitted, as required by law, but instead only looked for specific studies relating to flavors and youth use.
A memo dated July 9, 2021, written by Anne Radway, the associate director of the FDA’s Center for Tobacco Products’ Office of Science, states that “based on the information available to date, FDA has determined this evaluation requires evidence that can demonstrate whether an applicant’s new non-tobacco flavored product(s) will provide an incremental benefit to adult smokers relative to the applicant’s tobacco-flavored product(s). In particular, the evidence necessary for this evaluation would be provided by either a randomized controlled trial (RCT) or a longitudinal cohort study. The absence of these types of studies is considered a fatal flaw, meaning any application lacking this evidence will likely receive a marketing denial order.”
Radway goes onto explain that due to the large number of PMTAs received, the agency would only conduct a Fatal Flaw review of PMTAs for non-tobacco flavored ENDS products.
“The Fatal Flaw review is a simple review in which the reviewer examines the submission to identify whether or not it contains the necessary type of studies. The Fatal Flaw review will be limited to determining presence or absence of such studies; it will not evaluate the merits of the studies,” Radway states. “To decrease the number of PMTAs without final action by September 9, 2021, [Office of Science] used a database query to identify the top twelve manufacturers with the largest number of pending PMTAs [in the substantive review stage of the process] … Following completion of filing those applications that are filed will immediately initiate Fatal Flaw review.”
Radway also states that for the remaining PMTAs not in [substantive review] for non-tobacco flavored e-liquid products, FDA will send a “General Correspondence letter requesting the applicant to confirm if their PMTA contains such evidence and, if so, to direct FDA to the location in the application where the studies can be found.”
During the first day of TMA’s “From Chance to Change” webinar on Nov. 17, panelists were disturbed by the findings that the agency, rather than reviewing a submission on its merits, simply searched for the presence or absence of certain studies.
Brittany Cushman
Brittani Cushman, senior vice president, general counsel and secretary at Turning Point Brands said that the “idea that so many of the applications were reviewed with an eye toward this so-called fatal flaw analysis” didn’t “feel like the right direction” for the PMTA review process.
The FDA admitted it made an error in TPB’s PMTA review and TPB did in fact submit studies that the agency decided during the PMTA process were needed, after saying for years the studies were not required. The FDA then rescinded TPB’s MDO and placed its applications back into substantive review. The agency has since rescinded or a court has stayed MDO’s for 10 companies and the agency is currently facing at least 45 lawsuits for it handling of the PMTA process. This is in addition to the dozens of requests for supervisory review.
“The way the review process has played out this far, really, feels like the incentive structure in the nicotine industry has been placed on its head,” explained Cushman. “It seems that the lower-risk products are receiving heightened scrutiny, kind of an opaque direction as to what’s sufficient. And it just doesn’t feel like these products are getting a kind of equitable treatment in the space.“
Triton Distribution had their MDO stayed by the 5th Circuit Court of Appeals with the court holding that Triton is likely to succeed on the merits of its case because the FDA “changed its regulatory requirements” and that this “switcheroo” to now require a randomized controlled trial and/or a longitudinal cohort study – which the Agency previously stated on numerous occasions would not be required – was arbitrary and capricious under the Administrative Procedure Act.
The court stated that the FDA failed to “reasonably consider the relevant issues and reasonably explain” the MDO. The Court further noted that FDA failed to consider Triton’s marketing plan, surveys, and evidence of potential benefits of flavored e-cigarettes. FDA also “failed to consider the company’s legitimate reliance interests, as Triton relied on FDA’s statements made in numerous public meetings, guidance documents and rulemakings” that it did not expect applicants would need to conduct long-term studies to support their PMTAs.
Cushman told webinar watchers that, at the end of the day, the FDA’s regulatory treatment of the various product categories is to the detriment of the adult smoker.
“We’re all down in the weeds of this. But it’s difficult to see how we ended up at this point. And it certainly can’t be where anyone wanted this process to play out,” she said. “I think this has led to a lot of detrimental outcomes. You have adults seeing a large number of vapor products being deemed as not appropriate for the protection of public health while seeing no change in [combustible] cigarette offerings in their local C-store … This is being celebrated not only by those who are ignorant to the science, but more perversely, those [who understand the science and should] know better.”
For more on this session from TMA 2021 read the next issue of Vapor Voice coming in mid-December.
Synthetic nicotine could be considered a component of e-cigarettes, which would allow for the product to be regulated by the U.S. Food and Drug Administration. Mitch Zeller, director of the FDA’s Center for Tobacco Products, said the agency was concerned about the use of synthetic nicotine to avoid regulation and enforcement and is considering its options in dealing with its use.
Mitch Zeller, director of the FDA’s Center for Tobacco Products
On Nov. 17, the first day of TMA’s “From Chance to Change” webinar, Zeller said that the agency is charged with regulating tobacco products, which according to the Tobacco Control Act is anything that’s “made or derived from tobacco that is intended for human consumption, including any component, part or accessory of a tobacco product.” Zeller said that components and parts could include everything from coils and batteries to all the ingredients comprised in producing e-liquids (such as flavorings and vegetable glycerin) even if the product does not contain nicotine.
“That’s an assessment that we need to make on a case-by-case basis based upon the totality of all the information that we have,” said Zeller, adding that another challenge is that synthetic nicotine is now of such high quality and complexity that it has become difficult to differentiate it from nicotine derived from natural tobacco. “Historically, that hasn’t been a problem,” he said. “It’s not a problem now, but it could become a challenge for us going forward.”
Zeller explained that nicotine is comprised of two isomers: R and S. Tobacco-derived nicotine is 99 percent S, and early synthetic nicotine had a 50-50 split between R isomers and S isomers. However, newer versions of synthetic nicotine have much higher proportions of S isomers (as high as 99.9 percent pure), making it harder to tell synthetic apart from natural nicotine. Tobacco-derived nicotine is also becoming higher in quality.
“Tobacco-derived nicotine is now being made available at a higher quality … pharmaceutical grade from a purity standpoint. And with that, it may be harder for us to see that chemical fingerprint, if you will, whether it’s tobacco DNA or tobacco-specific nitrosamines,” he said. “We could see this as a problem going forward. Coupled with the clear intent of certain companies to do this to evade FDA regulation … We are concerned about what this means for product regulation, for the public health, and a product like Puff Bar proudly proclaiming its use of synthetic nicotine, [and] being the number-one brand used by youth.”
In the short term, Zeller said the FDA is talking internally about how to best address the growing number of products that are using synthetic nicotine to skirt FDA regulation. He said the agency is also responding to questions from Congress about synthetic nicotine and providing technical assistance to members when asked.
“There are a lot of companies out there that pride themselves on playing by the rules. They have every right to expect that the playing field is going to be level. That’s where we come in with our compliance and enforcement authorities,” Zeller said. “We agree that one of the most important things that we can do, using our compliance and enforcement tools, is to level the playing field and to have our actions [in the e-cigarette space], hopefully, serve as a deterrent. There’s nothing that I can say from a compliance enforcement standpoint on synthetic nicotine other than we have ongoing investigations.”
For more on Zeller’s speech at TMA read the next issue of Vapor Voice coming in mid-December.
A divided panel of the U.S. Court of Appeals for the Sixth Circuit rejected Breeze Smoke LLC’s application of a stay of the U.S. Food and Drug Administration’s order Friday, denying the company’s premarket tobacco product application (PMTA) for some of its vaping products.
In Breeze Smoke LLC v. FDA, the Sixth Circuit rejected the Fifth Circuit’s conclusion that the FDA had orchestrated a “surprise switcheroo” in the PMTA review process. This creates an interesting circuit split that might attract Supreme Court interest, according to Reason’s Jonathan Hadler.
The Sixth Circuit’s order, on behalf of Judges Moore and Gilman, concluded that the FDA had never committed itself to accepting PMTA applications for flavored vaping products that lacked long-term studies. Rather, the FDA had merely indicated that “it might accept evidence other than long-term studies, if that evidence had sufficient scientific underpinnings to meet the [Tobacco Control Act’s] statutory mandate of demonstrating that flavored ENDS devices are appropriate for the protection of public health” (emphasis in original).
Thus the court concluded that Breeze Smoke had failed to demonstrate the strong likelihood of success on the merits necessary to support a stay. Judge Kethledge dissented, noting his agreement with the Fifth Circuit’s decision in Wages and White Lion Investments LLC v USFDA.
While rejecting Breeze Smoke’s stay request, the Sixth Circuit panel did note some concern with the FDA’s handling of the company’s application, particularly its “formulaic consideration” of Breeze Smoke’s plans to prevent marketing to youth. This failing, and the impact of a PMTA denial on Breeze Smoke’s business were still not enough to convince a majority of the panel to enter a stay however.
Participants heard from scientists, retailers, legal experts and CTP Director Mitch Zeller.
Scientists, data analysts and legal experts shared their insights into the rapidly changing U.S. nicotine business on Nov. 17, the first day of TMA’s “From Chance to Change” webinar. Participants also heard from retailers and the industry regulator.
Mitch Zeller
Mitch Zeller, director of the Food and Drug Administration’s Center for Tobacco Products, reviewed the latest data on youth e-cigarette consumption, which he said continues to be concerning. However, Zeller was quick to point out that because the 2021 study was the first to be conducted completely during the Covid-19 pandemic, the data could not be compared to that of the previous year.
Zeller also provided an unprecedented behind-the-scenes peek into the center as it processed millions of premarket tobacco product applications. The agency received applications covering more than 6.5 million deemed products, and most of them were submitted close to the Sept. 9, 2021, deadline—a date that, Zeller reminded his audience, had been brought forward by a full year following litigation by public health groups.
Because companies were not required to submit their applications in a particular way, the agency had to be ready to process for a wide variety of formats. “We had to prepare operationally, technically and logistically to ‘ingest’ all those applications,” said Zeller, adding that the agency was thrilled its submission system did not collapse under the volume of last-minute applications.
The FDA has by now acted on the vast majority of applications, sending refuse-to-file letters, issuing marketing denial orders (MDOs) or, in a handful of cases, granting marketing orders. “We are down to 80,000 products—most of them in the final stages of review,” said Zeller. Those still-pending applications, he acknowledged, include ones submitted by the companies with the largest market shares because they tend to be the largest and most complex applications.
Zeller also commented on the rising popularity of synthetic nicotine, which some MDO recipients, including market-leading Puff Bar, have embraced as a tool to keep their products on the market because they believe it is outside of the regulator’s remit. The FDA defines a “tobacco product” as anything “made or derived from tobacco that is intended for human consumption, including any component, part or accessory of a tobacco product.”
Synthetic nicotine, said Zeller, presents a new challenge for the regulator, in part because it is increasingly difficult to distinguish the compound from its naturally derived counterpart. Nicotine, he explained, comprises two isomers: R and S. Tobacco-derived nicotine is 99 percent S, and early synthetic nicotine had a 50-50 split between R isomers and S isomers. However, newer versions of synthetic nicotine have much higher proportions of S isomers, making it harder to tell them apart from natural nicotine.
Jim Solyst
The first panel discussion of TMA’s online seminar, moderated by Jim Solyst, principal of JMS Scientific Engagement, debated the status quo from an applicant’s perspective. The panelists included Brittani Cushman, senior vice president, general counsel and secretary at Turning Point Brands; Beth Oliva, partner at Fox Rothschild; Brian Erkkila, director of regulatory science at Swedish Match; and John Pritchard, vice president of regulatory science at 22nd Century Group.
While all participants expressed appreciation for the FDA’s daunting workload, some voiced disappointment with the fact that many applications appear to have received only a perfunctory “fatal flaw” review—a review in which the agency, rather than reviewing a submission on its merits, simply looks for the presence or absence of certain studies. The panelists lamented that the pathway to market is more cumbersome for reduced-risk products than it is for deadly combustible products.
Participants worried also about how the public would interpret the lack of determinations on major applications, citing persistent misunderstanding of reduced-risk products and the continuum of risk by legislators, journalists and even physicians.
Asked to look forward, one panelist suggested the industry should consider what it would do when the next e-cigarette or vaping use-associated lung injury (EVALI) happens, referring to a mysterious outbreak of lung injuries in 2019 that was caused by illicit THC products but tainted the entire industry. Another participant stressed the importance of enforcement after all marketing applications have been decided. If any “yahoo” can sell products without authorization, she said, it would render the investments by the good actors worthless.
Mary Szarmach
The second panel of the TMA webinar, moderated by Mary Szarmach, senior vice president of governmental and external affairs at Smoker Friendly, reviewed the market from a retailers’ perspective. The panelists included Don Burke, senior vice president of Management Science Associates; Tom Briant, executive director and legal counsel at the National Association of Tobacco Outlets; and Amanda Wheeler, president of the American Vapor Manufacturers Association.
Burke sketched the latest trends in the nicotine market. The pandemic, he said, makes comparisons with 2020 difficult. With many people working from home last year, sales of cigarettes and large cigars experienced unusual growth, but as people returned to the office in 2021, those trends are starting to level off or are even reversing. Burke expects cigarettes to resume more normal consumption patterns next year. Modern oral continues its remarkable growth, albeit at a lower pace than last year because most convenience stores are by now carrying the product. And volume sales of vapor cartridges are up by more than 18 percent as the EVALI crisis fades from memory.
Briant provided a regulatory update, touching on the proposed nicotine tax hike in the Biden administration’s Build Back Better legislation, the FDA’s proposal to ban menthol in cigarettes and flavors in cigars and the status of graphic health warnings, which are currently being challenged in court. Litigation has pushed the implementation date to January 2023, and this could be further extended. Briant noted that there have been no hearings yet on the merits of graphic health warnings.
Asked to analyze vapor retailers’ current predicament, Wheeler drew an analogy with the Hindenburg disaster, after which shattered public confidence marked the abrupt end of the airship era. She cited the avalanche of MDOs, the U.S. Postal Service ban on shipping vapor products and the proposed federal excise tax on vapor products, which would make vapor products more expensive than some cigarettes.
Wheeler said these developments were driving vapers back to cigarettes, illicit producst and synthetics—many of them made abroad and falsely labeled. She described a “misguided crusade,” funded by deep-pocketed donors and cheered on by the irresponsible media. “When smoking was plummeting, they took action to make it increase; when American entrepreneurs innovated a news sector, they strangled it,” she said.
Asked what kept them up at night, the panelists named employee safety, flavor bans and lack of enforcement.
Szarmach related how a tax increase in Colorado had instantly resulted in more break-ins and robberies at her stores—an unwelcome development at a time when workers were already in short supply. Briant said that local flavor bans drove customers away without affecting total consumption—consumers would simply buy their products elsewhere. Wheeler said Arizona was not enforcing Tobacco-21 legislations, enabling bad actors to do good business.
The TMA online seminar continues today at 10:30 a.m. Eastern Time with a keynote from CTP Office of Science Director Matt Holman and panel discussions on “Your path to market” and global trends.
To register, please click here. Registrants will also have access to previously aired sessions.
U.S. President Joe Biden today announced that he would nominate Robert M. Califf, a former commissioner of the Food and Drug Administration, to lead the agency again, reports The New York Times.
A cardiologist and long-time consultant to drug companies, Califf ran the FDA during the last year of the Obama administration
“Dr. Califf is one of the most experienced clinical trialists in the country, and has the experience and expertise to lead the Food and Drug Administration during a critical time in our nation’s fight to put an end to the coronavirus pandemic,” Biden said in a statement.
Califf has been a forceful advocate for tobacco control; before he was the FDA commissioner, he was the agency’s deputy commissioner for medical products and tobacco. In an appearance with other former commissioners this year, he said, “I have never seen more capable or nastier lawyers than what I experienced in trying to deal with the tobacco industry. It was awesome and quite frightening for public health.”
After stepping down as the vice chancellor for clinical and translational health at Duke University, Califf has worked as a senior adviser to Verily Life Sciences, a health technology firm, and its sister company Google Health. Califf, who remains an adjunct professor of medicine at both Duke and Stanford University, is on the corporate board of Cytokinetics, a biopharmaceutical company, according to its website.
Califf said he was honored to be nominated for the position “at a critical time for our country,” adding, “There’s a lot of work to do, and if confirmed I look forward to rejoining the great team at the FDA to help in their inspiring mission to serve the public.”
The FDA has had seven different commissioners, including Califf, since 2012, when Margaret Hamburg left the post.
Confronted with an unexpected large volume of premarket tobacco applications, the U.S. Food and Drug Administration subjected some premarket tobacco product applications (PMTAs) to only a superficial review, according to documents obtained by Filter.
The PMTA review process comprises three phases: Phase I (Acceptance), which essentially means an application has been received; Phase II (Notification or Filing), which entails acknowledging a company had enough information for its applications to be formally filed; and Phase III (Review), which involves a substantive scientific evaluation, followed by a marketing granted order or marketing denial order (MDOs).
Overwhelmed by the large number of PMTAs and facing a court-ordered deadline of Sept. 9, 2021, the FDA in effect opted for a shortcut, according to Filter.
The publication cites a memorandum signed on July 9 by Matthew Holman, the director of FDA Center for Tobacco Products Office of Science (OS). “Considering the large number of applications that remain to be reviewed by the September 9, 2021, deadline, OS will conduct a Fatal Flaw review of PMTAs not in Phase III for non-tobacco-flavored ENDS products,” it reads.
The Fatal Flaw review is a simple review in which the reviewer examines the submission to identify whether or not it contains the necessary type of studies. “The Fatal Flaw review will be limited to determining presence or absence of such studies; it will not evaluate the merits of the studies,” the memorandum states.
Filter suggests that CTP reviewers created what’s probably a new method to get through a backlog of millions of PMTAs, searched those applications for longitudinal cohort studies and randomized clinical trials without evaluating any other evidence, and for applications lacking them, did not advance them beyond Phase II and just sent out templated MDOs.
The U.S. Food and Drug Administration devastates small businesses with a plethora of marketing denial orders.
By Timothy S. Donahue
At press time, the U.S. Food and Drug Administration had yet to approve an electronic nicotine-delivery system (ENDS) product for sale in the U.S. But it had killed much of the U.S. market for such products. As of Sept. 23, the agency had issued 323 marketing denial orders (MDOs) accounting for more than 1,167,000 flavored vaping products. In addition, the FDA previously refused to accept (RTA) or refused to file (RTF) a significant share of the nearly 7 million applications it received from more than 500 companies.
At least four lawsuits contesting MDOs have been filed in the 2nd, 4th, 6th and 11th Circuit Courts of Appeals against the FDA. Turning Point Brands (TPB) filed a petition for review with the United States Court of Appeals for the 6th Circuit. The petition forced the FDA to provide an administrative record for its decisions on PMTAs. TPB sells various flavored e-liquids marketed under the Solace, VaporFi and Vapor Shark brands.
In a surprise move as this magazine was going to press, the FDA rescinded Turning Point Brands’ MDO. The FDA admitted it made an error in TPB’s PMTA review and TPB did in fact submit studies that the agency decided during the PMTA process were needed, after saying for years the studies were not required. The FDA had not yet responded to the remaining cases as of press time.
“Upon further review of the administrative record, FDA found relevant information that was not adequately assessed,” the FDA letter to TPB states. “Specifically, your applications did contain randomized controlled trials comparing tobacco-flavored ENDS to flavored ENDS as well as several cross-sectional surveys evaluating patterns of use, likelihood of use, and perceptions in current smokers, current ENDS users, former tobacco users, and never users, which require further review.”
TPB was asking the court to review the FDA order “on the grounds that it is arbitrary and capricious, an abuse of discretion, contrary to the Federal Food, Drug and Cosmetic Act, as amended by the Family Smoking Prevention and Tobacco Control Act of 2009, and otherwise not in accordance with law.” The company requests the court “vacate or modify” the FDA order and asks that TPB be allowed to “continue to market the products subject to the challenged order.” Bidi Vapor filed a similar suit in the U.S. Court of Appeals for the 11th Circuit, BMF (Bad Modder Fogger) filed in the 4th Circuit and Magellan Technology, parent to DemandVape, has filed in the 2nd Circuit (those lawsuits are still active).
Credit: CASAA
In addition to its arbitrary claim, Magellan also claims in its court petition that the “FDA’s issuance of an MDO in the absence of a finalized rule” setting forth the required contents of a PMTA is unlawful. “FDA’s adoption of a comparative efficacy standard for the granting of a marketing order for non-tobacco- and non-menthol-flavored ENDS products versus tobacco-flavored ENDS products is, in reality, a disguised tobacco product standard that has been adopted and is being applied by FDA through adjudication rather than adopted through notice-and-comment rulemaking,” states Magellan’s petition.
According to Mitch Zeller, the director of the FDA’s Center for Tobacco Products (CTP), many of the accepted applications ultimately received an RTF letter because they did not include required information. “For example, companies received RTF letters for not including required content such as ingredient listings, labels for each product to be marketed or adequate environmental assessments,” he wrote.
In a joint news release with Zeller and acting FDA Commissioner Janet Woodcock, the FDA explained that the applications from many MDO recipients “lacked sufficient evidence that they have a benefit to adult smokers sufficient to overcome the public health threat posed by the levels of youth use” of ENDS products.
The PMTAs submitted by TPB and subsequently denied market access and the brought back under review by the FDA included an in-depth toxicological review, a clinical study and studies on patterns and likelihood of use, according to a motion to stay filed by TPB on Sept. 30. “In light of the unusual circumstances,” the FDA’s Center for Tobacco Products (CTP) Director Matt Holman stated in the letter. “FDA has no intention of initiating an enforcement action” against TPB’s products that had previously received an MDO.
Many of the current lawsuits against the FDA accuse the FDA of many of the same issues TPB’s withdrawn suit claimed. For example, TPB’s stay said the agency had moved the goalposts for data needed to receive a marketing order based on what the agency “learned” from the “review [of] PMTAs for flavored ENDS so far,” according to the stay. TPB noted that the “North Star of administrative law” is that agencies cannot induce regulated parties to rely on “agency representations about regulatory requirements” then penalize them using the previously unannounced criteria after the fact.
“But that is precisely what FDA did here,” the stay motion states. “[The] FDA reasoned that TPB failed to conduct ‘a randomized controlled trial and/or longitudinal cohort study’ or other studies performed ‘over time’ to show that TPB’s specific flavored products help adult users stop smoking more than tobacco-flavored products do. Yet FDA previously deemed these studies unnecessary.”
Tony Abboud, executive director of the Vapor Technology Association, suspects the FDA made an internal policy decision to change the PMTA standard to make it impossible after the fact for a company to comply and get a flavored ENDS application approved. “I think that that decision is being implemented application by application, which I don’t believe is fair under the law,” said Abboud. “I think that the refocusing on open system flavored e-liquids is a direct result of the public and political pressure that was placed upon the FDA by Congress, which expressly said they were trying to interfere with the regulatory process.”
What’s in a name?
Critics say the FDA has made several “sloppy” mistakes in reviewing PMTAs and issuing MDOs. Numerous companies say the agency was inconsistent in banning flavors based solely on the flavor’s name. Bidi Vapor’s parent, Kaival Brands, said that the agency banned its “Arctic” flavor, misidentifying it as a “not-menthol” flavor. TPB also says in its stay motion that the FDA is forcing TPB to pull nonflavored products from the market; however, the FDA’s order applies to “Authentic Tobacco” and “Bold Tobacco” yet not “Classic Tobacco” (which the FDA is still considering).
Credit: Cursed Senses
“Those are the same flavors with the same formulations; they just use different names across product lines. The same goes for ‘Ripe Tobacco’ (forbidden) and ‘Smooth Tobacco’ (reprieve) and for ‘Mint’ (banned) and ‘Mighty Menthol’ (allowed for now),” the stay explains. “It is anyone’s guess why some of these products must exit the market immediately yet others might pass muster if FDA actually reviews TPB’s studies.”
Since January 2021, the agency has issued at least 170 warning letters to firms that collectively have listed more than 17 million ENDS products with the FDA and that did not submit premarket tobacco product applications (PMTAs) for the products by Sept. 9, 2020. Applications for products manufactured by major companies, such as Vuse, Juul, Logic and blu, are still under review. During this time, the agency also granted substantial equivalence (SE) status (marketing approval) to over 350 combustible products from the cigar, pipe and hookah tobacco product categories.
Amanda Wheeler, president of the American Vapor Manufacturers Association (VMA) and the owner of Jvapes e-liquids (see “No Surrender,” page ?), assisted more than 230 small-sized to mid-sized e-liquid manufacturers in submitting PMTAs for more than 1.7 million products. Nearly all of those applications received either an RTA, RTF or an MDO.
Wheeler tweeted on Sept. 9 that it was a “tough day” for the industry because “lots of very good people who I respect deeply and who helped thousands of smokers quit got told by our government that their products were illegal. To all of you, I am so very sorry. To your customers, I am even more sorry. Our government is wrong on this.”
Before the announcement, many industry experts said that banning most e-cigarettes from the market could harm public health. In a commentary published on the Reason Foundation’s website, Guy Bentley, the organization’s director of consumer freedom research, states that the sooner that U.S. public health officials embrace vaping’s potential to improve public health by reducing smoking and smoking-related deaths, “the better off we’ll all be.” The result of shutting down a vast portion of the vape industry, he warns, will be more smoking.
Anti-vaping activists, by contrast, argued for a ban on e-cigarettes. In a recent blog post, Laurie Rubiner, executive vice president of domestic programs at the Campaign for Tobacco-Free Kids, and Linda Mendonca, president of the National Association of School Nurses and an assistant professor at the Rhode Island College School of Nursing, wrote that the “evidence is clear” that as long as any flavored e-cigarettes remain on the market, kids will get their hands on them (no reference to evidence was provided).
“To truly protect kids and end the youth e-cigarette epidemic, the FDA must eliminate the flavored and high-nicotine products—including the popular menthol flavor—that have driven this crisis,” the pair write. “Parents, educators and health advocates are counting on the FDA to take them off the shelves.”
Tom Miller, attorney general for the state of Iowa, said the FDA actions against flavors endanger public health. He said that the best science available indicates that most youths are not getting e-cigarettes from vape shops and that a significant number of adults are using products from vape shops to move away from combustible cigarettes.
“Let’s not forget the overwhelming risk to public health: The CDC [U.S. Centers for Disease Control and Prevention] estimates the burden of tobacco use in the United States is 480,000 lives a year, all of which is due to the use of cigarettes,” Miller said in a statement. “We believe in the strong, science-based regulation of alternative tobacco products, and the FDA is the best agency to undertake that task. Policymakers must strike the right balance between making accessible potentially lifesaving lower risk nicotine products while discouraging use by those who wouldn’t smoke, especially youth.”
Impacts of regulation
Several studies have suggested that if vape product sales were restricted to tobacco flavors, many would return to combustible tobacco. One study found that approximately one-third of U.S. vapers aged 18 to 34 say flavor bans would push them back to smoking traditional cigarettes. The study published in Nicotine & Tobacco Research analyzed data from February to May 2020 and looked at 2,159 young adults in Atlanta, Boston, Minneapolis, Oklahoma City, San Diego and Seattle, examining support for e-cigarette sales restrictions and the perceived impact of flavor and vaping bans.
Credit: JHVEPhoto
Two other recent studies showed similar results. A study in JAMA Pediatrics showed that following San Francisco’s flavor ban, teens were more likely to smoke than those in other school districts. A different study in Nicotine & Tobacco Research shows that teens who vape would be smoking cigarettes if vapes hadn’t become available.
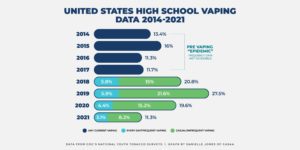
Recent evidence also seems to show that the overall youth use of e-cigarettes in the U.S. is declining. According to the 2021 National Youth Tobacco Survey (NYTS), the FDA and the CDC found that youth use of e-cigarettes fell sharply in 2021. It’s the second consecutive year of major declines. As is typical in the release of the NYTS data every year, media reports about the NYTS were all over the board. One headline read, “Big Drop in U.S. Teen Vaping with Covid Closures” while another read, “Teen Vaping Craze Shows No Sign of Slowing.”
The study shows that an estimated 11.3 percent (1.72 million) of high school students and an estimated 2.8 percent (320,000) of middle school students reported current e-cigarette use, lower than the 19.6 percent (high school) reported in 2020 and substantially lower than the 27.5 percent (high school) reported in 2019, according to previous FDA statements. Middle school vaping fell to 2.8 percent this year from 4.7 percent in 2020—a 40.4 percent decline. Middle school past 30-day vaping in 2020 fell 55.2 percent from 2019.
Chris Allen, chief scientific officer at Broughton, a contract research organization (CRO) delivering analytical, scientific and regulatory services for the ENDS industry, said that the FDA might well be using the NYTS to justify the “flurry of MDOs” issued for flavored e-liquids. He also said the majority of the companies that have fallen foul of the recent MDOs are responsible manufacturers supporting tobacco harm reduction.
“I completely accept that youth use is unacceptable; however, the issue doesn’t appear to lie primarily in open systems but a product that is currently outside the jurisdiction of FDA: a disposable containing synthetic nicotine,” Allen said. “Regardless of the product, or the source of nicotine, there’s no place for irresponsible marketing and distribution practices that keeps adding fuel to this fire. I fear that the latest action is simply going to lead to a seismic shift into the black market and unregulated (synthetic nicotine) products, which will be near on impossible for the U.S. government to control. From my personal perspective, this doesn’t seem an appropriate way to support THR [tobacco harm reduction].”
Industry representatives predict major battles at the state level. “States are just going to ban the sale of any non-FDA approved product,” said a vape shop owner, who asked not to be identified as he had not yet received an MDO. “This is just going to be a never-ending stream of court battles. I hope every company is at least considering appealing the MDO decisions. The whole PMTA process was a giant bait-and-switch.”
Manufacturers that submitted their applications by the Sept. 9, 2020, deadline but who have not yet received an MDO can effectively continue to sell their products as no ruling has been made on them; however, the FDA has made it clear that any company that does continue to sell these products will be doing so unlawfully, although they are not likely to face any enforcement action due to the agency’s limited resources.
Numerous companies are appealing their MDOs. Many are appealing MDOs they believe were wrongly issued because the PMTAs were for tobacco and/or menthol flavors. The AVM is helping its member companies file formal appeals with the FDA because the agency “in their sloppy haste, FDA not only threw out flavored products. They also threw out many [companies] [regular] tobacco and menthol flavors. We’re starting with some of those appeals specifically for what we feel were sort of administrative errors with tobacco and menthol and also working on broader appeals.”
Companies can also contact the CTP’s Office of Small Business Assistance (OSBA) with general questions regarding statutory and regulatory requirements, including the appeals process. Another option is the FDA Office of the Ombudsman, the agency’s “focal point for addressing complaints and assisting in resolving disputes between companies.”
Deanna Clark with the Clark-Esposito Law Firm stated in a blog post that each company must submit its own submission appealing the FDA decision. Companies should not send in an appeal combined with other companies, she cautioned. “Next, you want to address arguments refuting [the] FDA’s basis for your denial. It can’t just be where you’re complaining about how it’s unfair and the government sucks,” said Clark. “You need to use some rational basis behind what you’re submitting to them. And thirdly, you need to submit it to the right office and make sure it gets to the right people within the right timeframe.”
The e-cigarette saga with the FDA is far from over. Between lawsuits and appeals, many decisions may eventually be left out of the hands of the FDA entirely. The FDA’s ombudsman and appeals court judges could now decide the fate of flavored e-liquids. Congress could possibly step in and change the statutes, but many have said that is unlikely. The industry is also still waiting for decisions on the PMTAs filed by the major tobacco companies, and if anyone is approved, it may open the door for standard equivalency products. The only thing that hasn’t changed in the vaping industry is its uncertain future.
This year’s Global Tobacco and Nicotine Forum (GTNF) focused on innovation and sustainability in the ENDS industry.
By Vapor Voice staff
The Global Tobacco and Nicotine Forum (GTNF) has been one of the most insightful conferences over the past decade, especially in its embracing of electronic nicotine-delivery systems (ENDS) and their potential for harm reduction. During this year’s event, held in London from Sept. 21-23, speakers were challenged to focus their insights on this year’s theme: Continuing Change: Innovation & Sustainability. Below, we have provided session overviews of the many keynote speeches and panel discussions that centered on ENDS products. Next year’s GTNF will be held in Seoul, Korea on Sept. 20-22.
GTNF Fireside Chat with Todd Cecil, FDA
The U.S. FDA insists its banning of all flavored e-liquids other than tobacco is not a de facto ban on the products.
By VV staff
When the U.S. Food and Drug Administration began issuing marketing denial orders (MDOs) for vapor products, the industry was understandably shocked. Many companies that had submitted timely premarket tobacco product applications by Sept. 9, 2020, had expected to first receive a deficiency letter and not immediately an order to remove their products from the market. Some MDO recipients complained the agency had “moved the goalposts” by suddenly requiring studies that it had previously said were not required.
Credit: Malcolm Griffiths
At least four companies have filed lawsuits over their MDOs. All are accusing the agency of making “arbitrary” decisions and not reviewing the submitted data according to the statutes. In a “fireside chat” between Joe Murillo, chief regulatory officer for Juul Labs, and Todd Cecil, deputy director of the Office of Science for the FDA’s Center for Tobacco Products, during the recent GTNF in London, Cecil acknowledged the missing data that caused the flurry of MDOs is not required by the statutes that regulate tobacco products.
When asked what the “level of expectation” the FDA had in deciding whether to issue a deficiency letter or an MDO after a premarket tobacco product application (PMTA) was moved into scientific review, Cecil said that the agency followed “a randomized approach” to choose the applications the FDA would work on.
“The randomized approach identified a number of manufacturers’ products that went into this scientific review, and we … evaluated them from top to bottom,” he said. Cecil noted the agency began to see in some applications that “tended to have problems or missing materials that we needed in terms of benefits [of flavors]; that we learned we have to have that benefit piece … that evaluation that we spent several months working on taught us what we had to look for to be able to [conduct] a full scientific review.”
Cecil said that the agency just figured “if we know going right in that there are pieces missing, why will they go through a deficiency process and with a very short turnaround expecting to get back a full study that wasn’t completed previously?” So, instead of issuing a deficiency letter as required by statute, the FDA just handed out MDOs because the agency knew that it would take a company a significant amount of time and expense to conduct the new required longitudinal and cohort studies. Cecil was then asked why the agency filed the applications in the first place.
“We had to make a determinant how can we streamline this evaluation and determine those products that have at least the bare minimum for us to do a real and complete evaluation,” Cecil said. “This evaluation is not a standard. It is not a de facto standard or anything else. This is information that we need to see, but it’s not a requirement. An RTF [refuse to file] are those things that are required by the statute. And these studies are not necessarily required by the statute.”
The FDA has also been facing an unprecedented amount of scrutiny on its handling of the regulation of electronic nicotine-delivery system (ENDS) products and the PMTA process. Numerous health groups, anti-nicotine groups, states attorneys general and even members of Congress have criticized the FDA and demanded action. When asked if the FDA’s actions were influenced by these groups, Cecil said the agency focuses on science.
“We’re science-based,” he said. “We need look at what is presented to us in the application and in the laboratory. That is what we’re most focused on. If there is new data out there and that new data is brought to our attention through one of these [groups], then that’s fine. We would be happy to get those up and understand the bigger picture, all of the data … We need to evaluate those … scientifically or make a determination based upon that science.”
Only 100,000–200,000 products remain under FDA review. Of the 6.7 million submitted PMTAs, all others have received either a refuse-to-accept, an RTF or an MDO response. Cecil denied the agency was making a “categorical policy decision as opposed to an application-by-application decision” about flavored products. “We are stating that we understand that there is a significant youth initiation risk that comes from flavored ENDS products,” he said. “We are, in fact, reviewing all of those, and what we have found as we’ve done our reviews is that none of the literature is sufficient to demonstrate that there is not a youth initiation risk for individual flavors.
“We see that tobacco has a lower initiation risk. We see that menthol has some issues with it, and we are going to be evaluating that as we go forward. However, all of the data points to the flavored products as having significant youth initiation concerns. So what we’re looking for is an adequate indication that there’s a benefit on the other side of the equation. This is not a decision that we aren’t going to accept flavored products. Absolutely opposite. We need to ensure that there is concrete and robust data that demonstrates that there is an existing user benefit for those products.”
Cecil declined to say when more MDOs would be issued or when the agency would rule on major products, such as Juul, NJOY, blu and Vuse. “We continue to work diligently,” is all he would say. “There are a number of products that are well along. But no, I can’t tell you how many are those ones, but there are some that we’re hoping to move forward in the short term.”
The FDA’s recent action against flavored e-liquids does not mean that the FDA will never approve a flavored e-liquid, according to Cecil. He said that the rejected applications just lacked the required information that those products met the agency’s “appropriate for the protection of public health” standard. “You are welcome to reapply once you have addressed the issues that we provided to you,” he said. “And we will reevaluate that at a future date.”
GTNF Panel: The Fork in the Road: What is Next for Tobacco and Nicotine
Regulators must remember the vaping industry began with hobbyists and enthusiasts who built their own devices.
By VV staff
The vaping industry faces many challenges. The road to a viable future for these products must pass through sensible regulations based on science. In the current environment, unfortunately, this will be challenging, according to speakers on the GTNF plenary panel The Fork in the Road: What is Next for Tobacco and Nicotine. Misperceptions surrounding nicotine and vaping products, the panelists agreed, are furthered by the mass media’s “wonton disregard” for the science behind the tobacco harm reduction potential of electronic nicotine-delivery systems (ENDS).
Credit: Malcolm Griffiths
One speaker noted that in addition to many countries banning or erecting insurmountable barriers to vaping products, well-funded anti-nicotine activists are attacking the people who are bringing reduced-risk products to adult combustible cigarette smokers trying to quit smoking. These groups are opposed to the tobacco harm reduction that science and innovation can bring.
All of these activities together only serve to enhance the vaping industry’s problem: the massive public misperception that vaping is as deadly as smoking cigarettes. The fact that a significant number of physicians mistakenly belief that nicotine, rather than combustion, is responsible for smoking-related illness, bodes ill for the perceptions among the general population. “If physicians believe this, imagine the views of the average smoker in Kenya or Chicago, Illinois, or in Australia,” one speaker said.
While anti-nicotine activists have done their share to misperceptions, the vaping industry too is partly to blame, according to one panelist. The ENDS industry can do a lot more than feel helpless or complain, this speaker noted. Innovation in harm reduction cannot occur without the vaping industry’s support. That means responsible marketing, combating illicit trade, limiting youth access and making sure that the ENDS industry is doing what it can to prevent underage use.
Panelists also expressed concern about the direction of the vapor market in the wake of the U.S. Food and Drug Administration’s marketing denial orders (MDOs), with some describing a “Wild West” scenario. After receiving MDOs, some companies have turned to synthetic nicotine because that product currently is outside of the agency’s jurisdiction. A panelist said that the FDA’s “scorched earth” approach to flavored products is only creating bigger problems in the market, adding that if a market isn’t regulated, there is still going to be an unregulated illicit market that has the potential to be more deadly than that for combustible tobacco.
“Nobody wants kids to take up the products … it’s a very significant responsibility that we in industry be there to be the stewards of that concept in generating science and evidence,” a panelist said. “We should all be proud of the good science that is being generated … that is our responsibility: to generate and publish and participate in the scientific debate and pursue reasonable regulation. What is reasonable? I don’t know. It’s not going to be nothing. We all have to get over it and figure out what is the right way forward so we can go back to helping the consumer and making sure we’re only serving smokers who are looking for alternatives to combustibles.”
GTNF Panel: Consumers: The Key Stakeholders
Harm reduction should empower individuals to make their own choices about what products they consume.
By VV staff
For many people, the threats they face in day-to-day life are far more immediate than their long-term health. The mission of harm reduction should be to empower people to make their own choices about what products they consume and their own health decisions, even if those decisions don’t align with what public health experts would say is optimal. This was the general focus during a plenary panel discussion at the GTNF called Consumers: The Key Stakeholders.
Credit: Malcolm Griffiths
Most of the session centered consumers standing up and advocating for the industry, the global attacks on flavored e-liquids and growing threats from the World Health Organization (WHO), which remains suspicious of tobacco harm reduction. Panelists agreed that while some consumers prefer to remain on the sidelines, many others are willing to get organized and campaign for tobacco harm reduction and the vaping industry. “The consumer voice is very powerful,” a panelist said.
A major concern for the vaping industry is the concerted campaign against flavors. Flavors, according to one panelist, are used to by the industry’s enemies to redirect the conversation toward children. “They’ll say vaping flavors attracts children, and then they get us to play in their playground,” he said. “It’s very different. You [consumers] have got to keep asserting that adults use flavors.”
The WHO is a threat no matter what, the panel agreed. The global health body is now even talking about redefining smoke to include anything that’s heated and emits a vapor. “This means that any customizability of a product will be restricted and have limits on it, which basically means all the vape products will be the same,” explained one panelist. “These [recommendations] have to be resisted. The WHO doesn’t make laws, but it’s very influential, and these things can’t just be waved away.”
The scientific studies the WHO uses to justify its negative view toward next-generation products as tools for harm reduction are “fantasy and cherry-picked” studies, according to another speaker. “The people who are against harm reduction will never sleep. They’re always working, and they’re highly funded,” a panelist said. “[Consumers] have to stay alert, and they have to stay organized because, at the end of the day, there are more consumers than there are activists against harm reduction, and we’ll vote. So, consumers really have a big role to play.”
Consumers are the key stakeholders. However, when talking about consumers, regulators must acknowledge that not every smoker is the same, according to the panel. Many smokers don’t want to quit combustibles. “The important thing is to understand why and respect their choice,” a panelist said.
One speaker said that the industry also needs more responsible vape reviewers on YouTube because the current ones “are absolutely appalling.” The speaker urged consumers to make their voices heard in politics. “You’ve got to have somehow to get ahold of your Parliamentarians or your politicians in your country and get them to campaign on your behalf because there are many, many consumers, but you haven’t got great voice in government, and that’s what you really need to try and get,” he said.
At the end of the session, an audience member asked the panel if it could see a situation where consumers would sue regulators over counterproductive rules, such as flavor bans. “I have mentioned the fact that it would be interesting if someone could do a test case, but I don’t know whether that someone could come from the consumer side and sue [over regulatory action],” the panelist said. “It’s also expensive, and someone will end up having to pay if you lose.”
GTNF Panel: Science Driving Innovation
The nicotine delivery of products and being conscience of the environment are key points in innovation.
By VV staff
Regulators globally are becoming more understanding of what they expect next-generation tobacco products to accomplish. Regulators want manufacturers to demonstrate, on a product-specific basis, whether the vaping products are a benefit to combustible cigarette smokers. More importantly, manufacturers must ensure that vulnerable populations such as youth are not using these products.
Credit: splitov27
During the lunchtime GTNF panel “Science Driving Innovation,” one speaker also mentioned that manufacturers must be more conscious about the environmental impacts of vaping products too. The environment is a big issue in the minds of governments, regulators and society as a whole. The panelists agreed that vaping manufacturers should produce products that are environmentally sustainable.
“Think about all the batteries that go to waste every time an e-cigarette is disposed of. What are we doing as an industry to address the fundamental questions that society and regulators are concerned about?” a panelist asked. “We need to start thinking about what views of science we need to really put our investments in [and start] focusing on going into the future.”
Another major industry concern that should be addressed through innovation is youth initiation. One panelist said this topic should be a primary focus of scientific efforts relating to vaping products. Reduced-risk products must exist for adult smokers, so it’s imperative that the industry proactively addresses the underage use issue. “If we don’t, others will try to do it for us, and then collectively, we will all compromise the potential that [we are focusing on during the conference] today,” one panelist said. “It’s a critical balance. It’s important that we offer adult smokers an alternative, and we can also combat underage use. We can do both, and we must because there’s too much at stake if we don’t.”
Another speaker discussed her company’s global retailer compliance monitoring program. The company sends thousands of “mystery shoppers” into U.S. retail outlets that sell its vaping products and collects data around whether the retailers are abiding by federal age verification laws and/or other local policies.
“What we found is that retailers need help. There’s a lot going on in this world. We help them by providing information on how they’re performing, education and training, and we can also assist in changing their existing point-of-sale technology,” she said. “It can actually prompt the clerks to check ID when they’re selling an interesting new product. And it alleviates the mental burden on their end.”
Another concern for the industry that can be addressed through innovation is improving nicotine delivery and satisfaction. That satisfaction delivered by products today is not enough to sustain the large number of people we want to see switching from cigarettes to electronic nicotine-delivery systems.
“To achieve meaningful harm reduction, we need these products to appeal to and be affordable to most adult cigarette smokers. Which means those consumers would need to like the product and be able to afford the product,” a speaker said. “They need to be able to trust these products, and it requires a significant investment in innovation if you want to do it properly.”
GTNF Plenary Panel: Innovation as the Path to Progress
The more freedom the industry has to innovate, the more likely smokers are to transition away from combustibles.
By VV staff
Innovation is grounded in regulation. Regulators can either embrace innovation as a tool to support harm reduction, or they can regulate them to the point that any innovation is impossible to bring to market. During the GTNF panel Innovation as the Path to Progress, one speaker explained that the U.S. Tobacco Control Act was written with the goal that the state of public health will change over time. The idea is that as smokers quit and product standards are implemented, many may migrate to products lower on the risk continuum. As a result, as the state of public health changes, the products that the U.S. Food and Drug Administration determines to be appropriate for the protection of the public health (APPH) will also change.
Credit: Urupong
“If you think about the significance of the innovation of the e-cigarette, today we have major companies that are in the tobacco space talking about eliminating combustion altogether,” a panelist noted. “We have companies giving up their entire combustible segments, and that would not have happened, in my opinion, had it not been for the innovators.”
Making innovative progress in the vapor industry is measured by transitioning adult smokers to noncombustible products, according to the panel. However, there are many avenues to accomplish this goal as well as numerous obstacles. One speaker offered the audience three focus areas that he described as the pillars of innovation. The first pillar is product innovation. “If the product is not satisfying, people are not going to switch,” the speaker said. “In order to get there, we will need a very disciplined, science-based approach in understanding some of the questions underlying satisfaction. As we think about innovation and product innovation, it’s important for smokers to have a range of products to choose from.”
The second pillar is scientific innovation. There must be a comprehensive assessment of science to demonstrate that a product is APPH, and while all novel products tobacco products must be held to this high standard, it is rigorous and takes time. There are innovations in scientific methodologies that must be made, the speaker explained.
The speaker cited dissolution methods to understand nicotine release profiles and computation of toxicology as examples of tools that can help accelerate this pathway for getting products in the market. “Along with that, I think that regulators have an opportunity to create some innovative processes,” the speaker said. “For example, establishing product standards that will hopefully help these products be reviewed in an expedited manner, and most importantly, get them in the hands of consumers.”
The third pillar is communication. The industry needs to make clear the benefit to smokers by switching to noncombustible products. The industry needs to address the misperceptions surrounding nicotine and the wrong assumption nicotine causes cancer. “This clouds the decision-making process of adult smokers,” the speaker said. “As manufacturers in the U.S., we have to seek FDA authorization before we can communicate a modified-risk or modified-exposure order. That, too, is important but time-consuming and resource intensive. This is a responsibility for everybody to explore innovative communication approaches that can address these misperceptions.”
Another area ripe for innovation in the electronic nicotine-delivery system industry is environmental sustainability. For example, e-cigarette batteries contain heavy metals. The industry must innovate battery technology that will reduce their products’ environmental impact. Responsible disposal of any product is important. Regulation can also impact environmental issues. In the U.K., for example, requiring 10 mL bottles instead of larger bottles creates more waste.
Finally, synthetic nicotine also offers innovative advancements for next-generation products. “I think that when we talk about moving away from combustion, that is one thing, but when we talk about moving away from tobacco—in other words, giving consumers a truly tobacco-free option—that’s where science comes in,” a panelist explained. “The promise that is involved with synthetic nicotine is significant. They need to research it closely and recognize that it does provide certain benefits that perhaps the tobacco-derived nicotine does not.”
GTNF Keynote: Frank Han
The leader of FEELM said that exciting innovations are happening every day in the vaping industry.
By VV staff
During a keynote address for GTNF, Frank Han, senior vice president of Shenzhen SMOORE Technology Co. and CEO of its FEELM business division, said that there are exciting innovations—big and small—happening every day in the vaping industry. Vaping products using FEELM atomization technology have now reached millions of users in more than 50 countries.
Credit: Malcolm Griffiths
“Vaporization technology is still just at the beginning; we could welcome the opportunity for innovation to create a better life together … Basic Science Innovation has been the cornerstone for sustainable growth; it is the science of atomization that we need to build as the foundation supporting the industry,” he said, speaking through an interpreter. “As a firm believer of innovation, SMOORE has integrated disciplines like engineering thermodynamics and biomedical sciences into our atomization research.”
SMOORE has been actively learning to understand and assess the long-term health effects of vaping, according to Han. The company currently has seven research centers between the U.S. and China, “bringing in global talents” from different backgrounds. In addition to in-house R&D resources and efforts, SMOORE is also focused on partnering with leading universities to transform the company’s scientific discoveries into applied technologies. “The way vape products are manufactured is also constantly evolving; more effectively and definitely more environmentally friendly,” said Han.
SMOORE had begun operations using the first fully automated pod production line in the world. Each new manufacturing (single) line can produce 7,200 standard vaporizers per hour, double the previous generation’s output. “We have been working with business partners to improve sustainable practices in all stages of product development, especially manufacturing with the common goal of reducing carbon footprint,” said Han.
SMOORE is currently evaluating the underlying technology of atomization for its potential applications in other fields. “With one direction of our R&D focus on the atomization application in healthcare, I am proud that SMOORE has made progress on the research of atomized medication, along with partners from different sectors,” Han said. “The initial results are all positive. We are hoping in the near future, more and more people might be able to inhale medicines or even vaccines with atomization devices.”
Looking ahead at the vaping industry overall, Han said that policymakers and NGOs must be inclusive. Regulation has been a heated topic recently in both the U.S. and China, and while institutional innovations to promote healthy industry development and more balanced regulations are needed, regulators must also embrace vaping as a strategy to improve public health while safeguarding against youth initiation, he said.
“The global media must also be inclusive. We must value the media that report from an unbiased perspective, involving more people in the public dialogue on vaping, discussing the pros and cons and discovering the truth,” said Han. “I’d like to share an old Chinese saying here: ‘Though the road ahead is dangerous and difficult, we can only achieve our goals with constant efforts.’ We must press ahead with a sense of perseverance to expect a better future.”
Amanda Wheeler: NO SURRENDER
Her dreams and business crushed by a marketing denial order, Amanda Wheeler vows to continue the fight for vaping.
By Timothy S. Donahue
Amanda Wheeler got involved in the vapor business after a personal tragedy. Despite a cancer diagnosis at age 19, she was unable to quit smoking for another 11 years—until she discovered vapor products. Eager to share her success with others, Wheeler and her husband, Jourdan, opened JVapes, an e-liquid manufacturer and retail store in Prescott, Arizona, USA, in 2012.
Credit: Malcolm Griffiths
The business was successful, quickly expanding to multiple locations across three states. Wheeler was helping her customers quit smoking combustibles and became increasingly involved in advocacy. She joined several support organizations, including Arizona Smoke Free Business Alliance, Vaping Advocates of Oklahoma, Rocky Mountain Smoke Free Alliance, Smoke Free Alternatives Trade Association, American E-Liquid Manufacturing Standards Association and Vapor Technology Association.
In October of 2020, Wheeler and fellow business owner Char Owen created the American Vapor Manufacturers Association (AVM) to help small businesses navigate the U.S. Food and Drug Administration’s onerous premarket tobacco product application (PMTA) submission process. The organization also engaged in federal lobbying and sought to provide reduced-cost scientific testing and expert regulatory compliance advice to members preparing PMTAs.
Wheeler and the AVM assisted 230 e-liquid manufacturers that submitted PMTAs for more than 1.7 million products following a plan she developed with Azim Chowdhury, a partner with the law firm Keller and Heckman and a regulatory and public policy attorney with a focus on vapor, nicotine and tobacco product regulation.
The deadline for submitting PMTAs to the FDA was Sept. 9, 2020. Wheeler submitted timely applications and was allowed to keep her products on the market for up to one year while the FDA reviewed her submissions. The agency’s deadline to decide on all the applications was Sept. 9, 2021. When Wheeler’s application was accepted, she felt confident that her business could survive and that the industry had a future.
As the deadline approached, however, Wheeler became anxious. The FDA was slow to release information before the deadline. Then, on Sept. 9, 2021, Wheeler received a marketing denial order (MDO). The regulatory agency appeared determined to put the small company she and her husband had built, along with the industry she passionately defended, out of business.
That day, the FDA issued MDOs to more than 130 companies, requiring them to pull an estimated 946,000 products from the market. The bloodbath continued in the following weeks. At press time, the FDA had issued 323 MDOs accounting for more than 1,167,000 flavored electronic nicotine-delivery systems (ENDS). As of Sept. 28, not a single ENDS had been approved.
That’s how Wheeler found herself on the global stage, sharing her story with some of the largest players in the nicotine industry at the recent GTNF conference in London.
“[The] FDA knew that they didn’t have the time or the resources to give our products fair consideration, but instead of asking for help, they let the 9/9 deadline pass and left the more than 500 companies subject to their decision in an unstable and probably untenable position,” Wheeler explained. “The FDA’s arbitrary ruling effectively criminalizes thousands of long-standing businesses in communities all across the country. Those entrepreneurs now have to junk their inventory, fire their employees, stiff their investors, and defer their dreams.”
Wheeler said she was standing up for the “little guy”—the thousands of small business owners who manufacture, distribute and retail open system products in vape shops all over the United States. She explained that her business and other AVM members made every attempt within their means to comply with the FDA regulations. It was an expensive process. It was also a system designed for small businesses to fail from the very beginning, she said.
“My company personally submitted several hundred thousand pages of documents to the FDA in an attempt to comply with this one premarket tobacco application standard. The [FDA’s] decision doesn’t just make a mockery of that earnest work. It also makes the more than 10 million Americans who made the switch to vapor products—in our vape shops, with our liquids—into outlaws, too,” said Wheeler. “Their freedom as Americans no longer includes the right to use a product with none of the well-established, deadly effects of those other substances, and which has undoubtedly saved the lives of countless former smokers.”
Wheeler said the FDA, in an act of “regulatory arson,” was creating a tobacco-led monopoly over the vaping industry, as only the companies with the deepest pockets stand a chance to survive the agency’s cumbersome PMTA process.
She also focused on what she perceived to be one of the biggest challenges facing the industry today: misinformation. “There is one other group I want to address with my time here. It’s the activists and the press who—whether because they are misguided or malicious—spread the falsehoods and distortions that directly led to this tragic outcome,” she said. “In this malign effort, those activists had enthusiastic help from nearly the whole of the national news media. By focusing on the messaging of Bloomberg dark money NGOs [nongovernmental organizations] and beneficiaries of MSA funds, our media and political class have criminally neglected the harm reduction aspects of vaping under the guise of moral virtue. The years added to their lives by our products are never mentioned.”
The misinformation plaguing the vapor industry has been around since Hon Lik introduced e-cigarettes to a mass market in 2006. TheWall Street Journal, for example, recently ran a gushing story about a Truth Initiative advertising campaign that misleadingly asserts that vaping nicotine “can worsen symptoms of anxiety and depression.” There is no evidence to support the claim. According to Wheeler, the statement is also contradicted by studies on the Truth Initiative’s own website. The Journal article even quoted a Truth Initiative executive admitting that “it is unknown whether a causal link exists” between nicotine and those symptoms.
“Just last month, FDA records gathered by Freedom of Information Act laws revealed that America’s most preeminent news organization, the New York Times, would send its articles in their entirety and before publication, to FDA officials for review and feedback. Neither that reporter, Sabrina Tavernise, nor her editors have summoned the integrity to offer any explanation,” Wheeler said. “Remember, these are publications and outlets that routinely praised and awarded themselves for taking on Big Tobacco. And yet on a decision that has given Big Tobacco exactly what they wanted—a monopoly—they are silent. Marching arm in arm with the very businesses they once excoriated as merchants of death.”
The biggest victims of the FDA’s actions, according to Wheeler, are the vapers who will now struggle to acquire the products that have helped them stay off of cigarettes. Wheeler vowed she would continue to fight for her customers and fellow business owners. “Even through their dismay, I am hearing a constant refrain: We are not going to stand for it,” she noted.
“We will be at the FDA’s doorstep demanding answers or forcing them through Freedom of Information Act laws and the courts. We are not surrendering our business or abandoning vapers to cigarettes,” she said. “As we say in Arizona, this is more than just a fight. It’s going to be a reckoning.”
Wages and White Lion Investments, parent to Triton Distribution, has been granted a stay of the marketing denial order (MDO) it received from the U.S. Food and Drug Administration. The panel of judges for the U.S. Court of Appeals for the Fifth Circuit issued the order on Oct. 15 that also granted motions to expedite the appeal case and a ruling for emergency relief.
Credit: Pixelbliss
The motion granted means the company can continue to market its electronic nicotine-delivery system (ENDS) products until the court decides on the company’s appeal of the FDA’s decision to deny its premarket tobacco product applications (PMTAs).
Triton Distribution filed a motion to stay after the FDA denied the company’s PMTA, in which Triton stated that it had been irreparably harmed as a result of the FDA’s actions and faced an imminent shutdown of its business if the motion to stay had not been granted.
“Black-letter rules of administrative law prevent an agency from retroactively changing legal requirements and from doing so without accounting for reliance interests. FDA failed to satisfy these requirements when it executed an about-face on the evidence it required to support a premarket tobacco product application (“PMTA”) for a marketing order for flavored electronic nicotine delivery system (“ENDS”) products almost a year after such applications were due,” the motion states. “FDA also acted arbitrarily and capriciously by ignoring relevant evidence found in Petitioner Wages and White Lion Investments, LLC d/b/a Triton Distributions (“Triton”) PMTA and applying a double standard to its consideration of that evidence when it issued Triton a marketing denial order (“MDO”). Further, by imposing a new, across-the-board requirement that flavored ENDS products be demonstrably more effective at promoting smoking cessation than otherwise identical tobacco-flavored products, FDA acted contrary to its authority under Section 910 of the Food, Drug and Cosmetic Act (“FDCA), 21 U.S.C. § 387j, and not in accordance with law.”
At least six companies have filed lawsuits challenging the agency’s decision to make the companies remove their products from the market. Last week, the FDA rescinded the MDO issued to Turning Point Brands (TPB) and the company will be allowed to continue marketing its vapor products while the FDA re-reviews the company’s premarket tobacco product application (PMTA).
The FDA admitted it made an error in TPB’s PMTA review and TPB did in fact submit studies that the agency decided during the PMTA process were needed, after saying for years the studies were not required. “Upon further review of the administrative record, FDA found relevant information that was not adequately assessed,” reads the FDA letter to TPB. “Specifically your applications did contain randomized controlled trials comparing tobacco-flavored ENDS to flavored ENDS as well as several cross-sectional surveys evaluating patterns of use, likelihood of use, and perceptions in current smokers, current ENDS users, former tobacco users, and never users, which require further review.”
Former U.S. Food and Drug Administration Commissioner Robert Califf is expected to return to the regulatory agency after President Joe Biden nominated the cardiologist for the position. According to the Washington Post, if confirmed by the Senate, Califf would take over an agency poised to make key decisions on the vaping industry, as well as coronavirus vaccines and treatments while facing criticism of recent controversial drug approvals and widespread burnout due to the ongoing pandemic.
Robert Califf/ Credit: Duke University
Califf previously served as commissioner for nearly a year in Obama’s second administration after an overwhelming vote in his favor. The White House has not finalized its decision, and the people with knowledge cautioned the situation could still change. But nine months into its search for a permanent FDA chief, Califf is now viewed as the leading candidate for the job.
Despite the agency’s prominent role in the nation’s coronavirus response, it has been without a permanent leader since Biden took office in January. Califf would replace acting Commissioner Janet Woodcock, the agency’s longtime drug chief, who has helmed the agency for nine months. Califf returned to cardiology after his tenure at FDA and currently works for the Duke Clinical Research Institute, but also took a position leading health policy at Google’s parent company Alphabet in 2019.
While he garnered support from many senators in his 2016 confirmation process, Sen. Bernie Sanders (I-Vt.) voiced concerns about his industry ties and said he could oppose the vote. Califf at the time had written papers with pharmaceutical industry executives and consulted for drug and device makers.