Today, the U.S. Food and Drug Administration (FDA) issued 10 warning letters to retailers and manufacturers who sell, manufacture and/or import unauthorized electronic nicotine delivery system (ENDS) products targeted to youth or likely to promote use by youth.
The warning letters were sent to establishments marketing unauthorized products, such as a backpack and sweatshirt designed with stealth pockets to hold and conceal an e-cigarette, ENDS products that resemble smartwatches, or devices appearing as children’s toys such as a portable video game system or fidget spinner.
Warning letters were also issued to companies marketing e-liquids that imitate packaging for food products that often are marketed and appeal to youth, such as candy, or feature cartoon characters like SpongeBob SquarePants.
“The FDA is focused on manufacturers and retailers that make and sell ENDS products that are targeted to youth and increase their appeal. The public should really be outraged by these products. The FDA is especially disturbed by some of these new products being marketed to children and teens by promoting the ease with which they can be used to conceal product use, which appeals to kids because it allows them to conceal tobacco product use from parents, teachers, law enforcement or other adults,” said Mitch Zeller, director of the FDA’s Center for Tobacco Products. “Even in the midst of the COVID-19 pandemic, we have not lost our focus on protecting youth against the dangers of e-cigarettes and will do everything we can to take action. These warning letters should send a clear message to all tobacco product manufacturers and retailers that the FDA is keeping a close watch on the marketplace. If you’re marketing or selling these products to youth, the FDA will not tolerate it.”
The following retailers and/or manufacturers or importers received a warning letter:
Shenzhen Uwell Technology Co., Ltd. d/b/a DTD Distribution Inc. (importer, retailer)
The FDA has also issued warning letters to 73 brick-and-mortar retailers for selling unauthorized flavored, cartridge-based ENDS products. This follows 22 warning letters that FDA issued last month for similar violations to online and brick-and-mortar retailers and manufacturers across the country. These warning letters are part of a series of ongoing actions consistent with the FDA’s recently issued policy of enforcement priorities for e-cigarettes and other deemed products on the market.
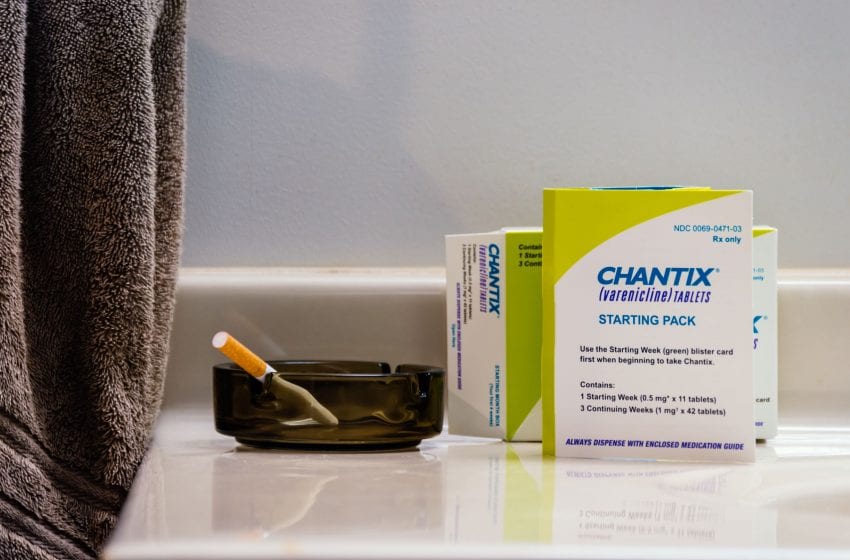
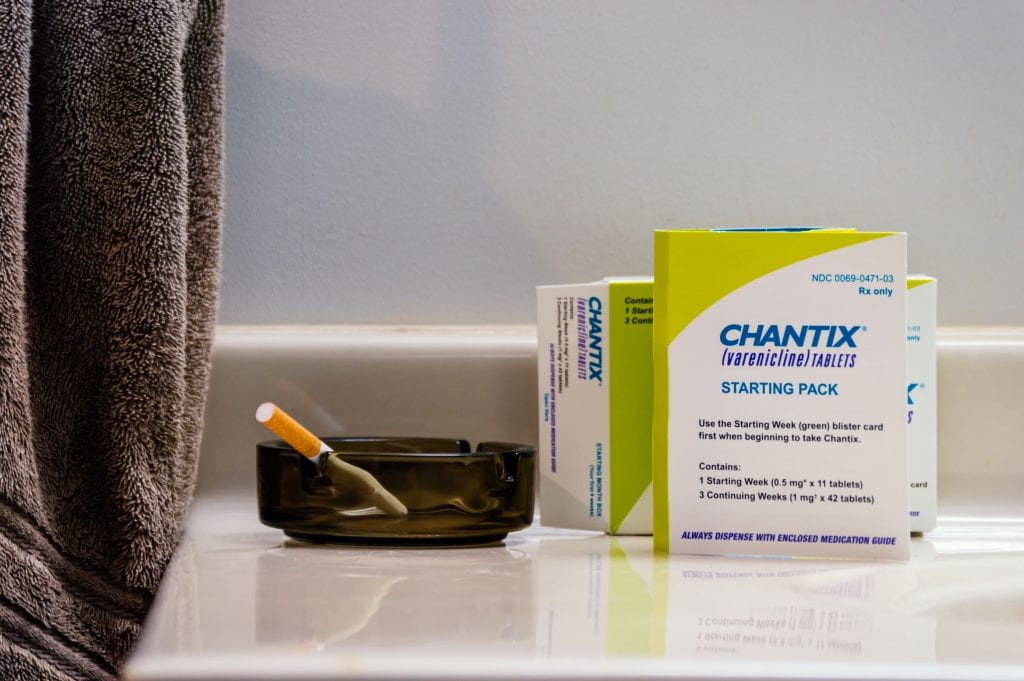
Doctors often recommend Chantix to their patients who smoke, but the drug has a long history of serious side effects.
By Maria Verven
Smokers have always been at a higher risk of heart and lung disease, but the Covid-19 pandemic has made quitting combustible cigarettes even more urgent. Cigarette smoking is a cause for concern because smoking inhibits the body’s ability to heal from infections such as the coronavirus, says Michael Siegel, a Boston University School of Public Health professor with more than 30 years’ experience in tobacco control, including two years at the U.S. Centers for Disease Control and Prevention (CDC) Office on Smoking and Health.
While the U.S. Food and Drug Administration (FDA), the CDC and the media often denigrate and vilify e-cigarettes, the drug varenicline, sold as Chantix, continues to fly relatively smoothly under the radar screen. When searching Google for e-cigarettes, you’ll get a whopping 386,000 results, including the predictable articles about youth e-cigarette use. In contrast, a search for Chantix nets only about 7,000 results, including a new study (read on to learn more) the authors use to promote its supposed safety and support the FDA decision to remove its black box warning.
Search on the CDC site for Chantix and you’ll get only 66 results versus over 2,200 for e-cigarettes; the top results for e-cigarettes mention “pulmonary disease,” “lung injury” and “youth tobacco use” even though e-cigarettes contain no tobacco. In contrast to nearly 3,300 results for the search term e-cigarette, the FDA site also only lists 78 results in a search for Chantix, and the top result is an update on their September 2016 decision to remove the boxed warning based on the conclusion that the “risk of serious side effects on mood, behavior or thinking is lower than previously suspected.”
Antwon Mcmullen I Dreamstime.com
BLOCK BOX WARNS OF SUICIDE AND PSYCHIATRIC PROBLEMS
Approved under the brand name Chantix in 2006 for smoking cessation, the drug varenicline originally carried a label warning consumers of the risk of suicide and other psychiatric problems, including depression and mania, psychosis, hallucinations, paranoia, delusions, homicidal ideation, aggression, hostility, agitation, anxiety and panic as well as suicide and suicidal ideation and attempt.
By the end of 2007, the Institute for Safe Medicine Practices (ISMP), a nonprofit medication watchdog, reported that Chantix was responsible for more reports of serious drug adverse events than any other drug in the U.S. “Data show that varenicline, ‘Chantix,’ continues to account for more cases of suicidal, self-injurious or homicidal thoughts than any other therapeutic drug from 2007 through 2013 Q3—more than three times as many as the second-ranked drug,” the ISMP reported.
Four leading nonprofit consumer, research and medical organizations—Consumer Reports, , the National Center for Health Research, the National Physicians Alliance and Public Citizen—joined the ISMP in filing a citizen petition with the FDA, documenting the extensive scientific evidence of Chantix’s substantial risks.
The FDA finally added its strongest black box warning in July 2009 after receiving 153 reports from doctors who were deeply concerned when their patients suddenly started exhibiting suicidal behavior.
By the fall of 2013, Chantix was associated with 2,748 adverse events (14.8 percent of all cases), including 293 suicides, 490 attempted suicides and many other cases involving self-injurious behavior or homicidal ideation—more than any other medication sold in the U.S. The ISMP ultimately recommended that “patients and doctors exercise caution in the use of varenicline and consider the use of alternative approaches to smoking cessation.”
CLASS-ACTION LAWSUIT FLIES UNDER THE RADAR
Not surprisingly, these adverse events spurred thousands of lawsuits by families of loved ones who suddenly and inexplicably took their lives while on Chantix. In March 2013, Pfizer, the drug’s maker, settled 80 percent of these lawsuits out of court for an estimated $273 million.
What’s odd is that any reports of adverse events associated with Chantix end then and there. Try searching for how and when the other 20 percent of lawsuits have been resolved or the latest statistics on how many attempted or successful suicides or homicides have taken place over the last six years, and the trail runs cold.
However, it’s easy to uncover the fact that Chantix has been linked with a host of neurologic or cardiac risks, including blackouts, convulsions and impaired vision. Due to these and other well-known psychological side effects, the Federal Aviation Administration banned pilots and air traffic controllers from using Chantix; the Department of Defense and the Department of Transportation also disallow its use by missile crews, armed military personnel, police, fire and emergency professionals and anyone in a “sensitive” occupation.
Yet it recently became even easier to buy Chantix—as of 2019, at least 12 states now enable consumers to purchase the drug without a doctor’s prescription. Between 2013 and 2018, the cash price for Chantix increased by 106 percent. Pfizer reported that U.S. sales were $899 million in 2019, up 7 percent from 2018. But another development may make the drug even more accessible and affordable: Pfizer’s patent protection expires in May 2020, so a generic version could potentially be available as early as this June.
Not surprisingly, Pfizer is seeking a court order to block copies of its drug until all its patents have expired—including those set to expire in November 2020, November 2022 and February 2023.
SUSPICIOUS STUDIES
Back in September 2016 when the FDA’s advisory committee met to review the black box label on Chantix, the meeting got quite contentious. There appeared to be much disagreement over whether to remove the boxed warning. Indeed, of the 19-member panel, five actually recommended strengthening the label and four recommended a language change.
But most suspicious was the evidence used to remove the black box warning: The voting members of the panel were presented with an April 2016 clinical trial paid for by Pfizer. Even the FDA noted a number of violations in Pfizer’s clinical trial protocols, including inconsistencies in source data, missing documents and safety assessments, and failure to record adverse events.
Plus, the personnel who performed the diagnostic interviews and mental health evaluations failed to meet the most basic professional qualifications. In fact, six of the 10 authors worked for either Pfizer or GlaxoSmithKline, maker of Zyban, a brand of antidepressant bupropion that is used as a quit smoking aid, while three, including the lead and second-named authors, were regular consultants paid to promote Chantix. The study was so poorly done that the FDA’s Office of Scientific Investigation determined that “it was neither feasible nor possible to attempt to adjudicate the cases based on the provided information,” concluding that “[Pfizer’s] investigator assessment of relatedness has been altogether disregarded.”
A more recent study reported in February 2020 in Addiction Magazine sought to assess Chantix’s relative cardiovascular and neuropsychiatric adverse effects in adults with no history of depression. Lo and behold, the retrospective study of over 600,000 adults found there was a 20 percent lower risk of of developing cardiovascular and neuropsychiatric adverse effects using Chantix when compared with the use of nicotine-replacement therapies (NRTs) such as nicotine patches and gums, and a 35 percent lower one-year risk of neuropsychiatric hospitalization when compared to patients using NRTs.
Interestingly, NRT was chosen for the comparison group “because it is the oldest, least expensive treatment, often available without prescription,” according to the authors. However, of the quit methods used, 35 percent of the subjects had substituted some regular cigarettes with e-cigarettes, and 24 percent had switched completely to e-cigarettes (a total of 60 percent) while only 25 percent used nicotine patches or gums.
The most obvious problem with this study is that it excluded anyone with a history of depression. Indeed, even the authors had to include these caveats: “The exclusion of patients with a diagnosis or treatment for depression limits the external validity of our results.”
“This study does not necessary demonstrate the safety of Chantix among people with baseline depression,” said Siegel. “It is possible that the suicidal ideation that seemed to be precipitated by Chantix in some cases occurs specifically among people with pre-existing depression.”
And one must question why the authors chose to compare Chantix with less popular quit methods while neglecting to include comparisons with e-cigarettes. “It only compared Chantix with NRT therapies but excluded the millions of people who have switched to e-cigarettes. The comparison would have been enlightening,” Siegel said.
WHICH METHOD HELPS SMOKERS QUIT?
Has Chantix been effective in helping people quit smoking? A study reported in the National Institutes of Health, called “Diminishing benefit of smoking cessation medications,” found that only 18.7 percent of people treated with Chantix remained abstinent from smoking after one year.
Even NRT, notoriously ineffective for long-term smoking cessation, was more effective. “There is no clinical trial directly comparing e-cigarettes with Chantix,” Siegel said. However, he cited a clinical trial published in the New England Journal of Medicine last year that reported a 20 percent quit rate with e-cigarettes—a similar rate to Chantix. “I would say that the best evidence at the current time suggests that e-cigarettes and Chantix are probably comparable in their efficacy in helping people quit smoking,” he said.
While there are no studies comparing Chantix and e-cigarettes, a study published in February 2019 in the New England Journal of Medicine that was funded by the National Institute for Health Research and Cancer Research U.K. compared e-cigarettes with NRT. Of the 886 participants, 18 percent who used e-cigarettes achieved abstinence from smoking one year later compared with only 9.9 percent in the NRT group.
Their conclusion? “E-cigarettes were more effective for smoking cessation than nicotine-replacement therapy when both products were accompanied by behavioral support. Switching completely from cigarette smoking to e-cigarette use would be expected to reduce risks to health.”
Unfortunately, it may take a long time before physicians and the public health community recommend e-cigarettes over Chantix to patients who want to quit smoking. The American Lung Association launched a “Quit, Don’t Switch” campaign earlier this year. Drawing a hard line that ignores the potential harm reduction offered by NRT and e-cigarettes, the site states, “Switching to e-cigarettes does not mean quitting. Quitting means ending your addiction to nicotine.”
“I don’t think physicians will widely embrace e-cigarettes for smoking cessation until the FDA has officially approved these products and the major anti-smoking groups have also embraced these products,” Siegel said. “Although the FDA will eventually approve some e-cigarettes, I don’t see the major anti-smoking groups embracing e-cigarettes any time soon.”
“I think Chantix should be recommended by public health officials as a Plan E smoking cessation remedy after cold turkey, e-cigarettes, smokeless tobacco and NRT, since Chantix poses far greater risks than these four other smoking cessation methods,” said Bill Godshall, founder and executive director of Smokefree Pennsylvania.
“E-cigarettes should be recommended to smokers who are unable or unwilling to quit by themselves or with Chantix or any other method—and that’s the majority of smokers,” said Konstantinos Farsalinos, a cardiologist, clinical researcher with the Onassis Cardiac Surgery Center in Athens and often-quoted expert on e-cigarettes.
“In a real-world setting, I think it is important to approach smoking cessation on an individual level beyond the results of any randomized trials. But most doctors discourage e-cigarette use to those who are unable or unwilling to quit with Chantix,” he said. “Even worse, they discourage vapers who have already quit smoking from using e-cigarettes. The end result is that many of them relapse to smoking.”
Maria Verven
The original “Vaping Vamp,” Maria Verven owns Verve Communications, a PR and marketing firm specializing in the vapor industry.
After a brief shutdown of all operations, vapor hardware manufacturers in China are now operating at more than 80 percent of pre pandemic production.
By Timothy S. Donahue
It’s not the same as it was this time last year. However, vapor hardware manufacturers in China that produce products for the world market are back in service after a brief hiatus due to the Covid-19 pandemic. Mostly based in Shenzhen, the e-cigarette capital of the world, companies say that they are working hard at implementing new standards and processes in order to keep employees and customers safe.
They are also playing catch-up in collecting data for premarket tobacco product authorizations (PMTA) in the U.S. market. “The coronavirus is indeed having an impact on the PMTA process,” says Welford Ou, CEO of Smoktech, a major manufacturer. “For example, the behavioral investigations have been stopped, and it is also taking more time for us to prepare all the products for the PMTA.” As of this writing, PMTA applications are due to the U.S. Food and Drug Administration (FDA) on May 12.
Smoore Technologies, the parent to Vaporesso and Feelm, says it “took strong and comprehensive measures” in advance of the virus’ outbreak. “We set up disease prevention and control teams in each of our facilities before the Chinese Spring Festival holiday. Dating back to the 20th of January, Feelm teams started to collect information, investigate employees’ conditions, prepare epidemic prevention supplies and disinfect public areas,” said Sofia Luo, marketing director for Feelm. “Before getting back on track, Feelm handed out a Covid-19 prevention and control booklet to each employee, providing scientific support to enhance health security.”
All Smoore facilities are now back up to at least 85 percent of pre-pandemic production, according to Luo. She says Feelm is already shipping goods to the U.S. and has been since the middle of February. “Smoore and its subsidiaries have enough key materials inventory, and all of our supply chain has recovered and is back in production,” she said. When asked whether there was a supply shortage, Luo replied, “In general, the impact of supply shortage is under control.”
Smoktech is getting back to its normal operations and more workers are expected to be hired as the virus outbreak is brought under control, according to Ou, who added that the company is awaiting the return of some workers from Wuhan (located in the Hubei province and the epicenter of the pandemic) where the quarantine was officially lifted on April 8.
“The sales are doing well even with the Spring Festival and a long time staying at home for virus control,” says Ou. “Our challenge is to get more skilled workers in [a] short amount of time to meet the growing demand. My concern for the world market is [that] hopefully they will embrace vaping and see it is better and safer than smoking cigarettes.”
Smoore took early action in order to prevent a massive global disruption in the vapor and e-cigarette market, according to Luo. She said that the company started to communicate frequently with its clients at the early stages of the pandemic. “We reminded customers to pay great attention to the epidemic. With the development of Covid-19 worldwide, most of our customers have been prepared (in terms of staff safety and inventory),” Luo stated. “The [virus] was a black swan event of great magnitude.” A “black swan” event refers to an unforeseen occurrence that typically has extreme consequences; in contrast, a “gray rhino” event is an obvious yet ignored threat.
Luo says that, although Covid-19 is spreading all over the world, the demand for e-cigarettes hasn’t changed. “Vapor stores in many countries remain open. And we know some brands are increasing the online sales and e commerce service,” she said. “We forecast [that] the 2020 industry will be the same size or a slight increase compared with 2019. And after PMTA, [the industry] will get a big increase.”
When it comes to issues like the limitation of freight and whether the virus could be on packaging, Feelm says the information from official sources are that there’s no limitation of freight, and the World Health Organization (WHO) confirms coronaviruses do not survive for extended periods on objects such as letters or packages and that it’s safe for people to receive packages from China.
Luo says Feelm will continue to stay updated on related information and adds that the company will spare no efforts to guarantee each product is clean and safe in all aspects from purchasing raw materials to exporting goods. Feelm products have been approved by several international quality and safety systems, according to Luo. “As a leading company in the automatization area, Feelm has the strength to embrace health and security first,” she says. “Disease prevention and control is our responsibility. Feelm is well prepared to protect employees’ safety to recover production and to win the battle.”
At its factories, Smoore and its other entities’ staff are well-equipped with protection products such as masks, gloves and other safety equipment, according to Luo. “Disinfection in public areas is proceeded twice a day. Exclusive dust bins for used face masks are placed throughout the facilities,” explains Luo. “All these measures lay a solid foundation to protect Feelm and all Smoore employees’ safety. This allows for the greatest possibility for full production recovery.”
THE NEW NORMAL
United we stand, together we win. Since the outbreak of Covid-19 at the end of last year, the Chinese government has implemented a series of powerful measures that have effectively controlled the spread of the virus, according to Sofia Luo, marketing director for Feelm, a major atomizer company based in Shenzhen, China.
“At present, production recovery has become the top priority. Allied with government regulations and a corporate plan, Feelm employees started to work online at the beginning of February. Now, all facilities serving Feelm clients are getting back on track,” explains Luo. “Thanks to Feelm’s advantages with having a strong supply chain and the ability to deal with emergencies, Covid-19 has had little impact on production. Once getting back to work, the production capacity will recover soon.”
In order to ensure the prevention of a recurrence of the virus within the company, Feelm has implemented an eight-step policy to enhance protocols and address health and security concerns for its employees. According to Luo, the first step requires all employees to register their current health status and any other conditions they may have.
“They must also inform us of the dates they are returning/returned to Shenzhen, the people they have met, etc.,” she says. “They must archive related documents setting up an exceptional first line of defense. Step two: any employees returning to Shenzhen from other places must isolate themselves at home for 14 days. They must work from home or online if applicable, according to Luo. Only if without any suspected symptoms like fever and coughing, are they permitted to return to the office. All employees must wear a face mask at [the] factory.”
Step three is all factories must have epidemic prevention supplies ready, according to Luo. Employees must wear a face mask while going out and have their temperature taken before stepping into the factory. They must also wash their hands after touching anything and always have disinfectants at hand.
Step four involves having strict Allied Social Sciences Association (ASSA) access control systems in place. Employees must wear identification badges and have their temperature taken twice a day. “Anyone whose body temperature is over 37.3 degrees Celsius should stay at home for medical observation,” says Luo. “Protecting yourself is protecting others.”
Luo explains that step five is one of the most vital steps. She says the company actively disinfects all public areas in factories twice a day. “Office areas are disinfected once a day after work. Public space and dormitories are also once a day,” she says. “Production area disinfection is arranged by departments.”
Step six involves maintaining the cleanliness in production workshops. “Enter the air shower room after disinfection, no more than six persons at one time. [The] distance between two workstations should be wider than one meter,” says Luo. “Reduce the number of employees at one production line. Add extra production lines if necessary. Spare no efforts to guarantee production safety.”
The final two steps are more lifestyle changes, according to Luo. Step seven centers on employees keeping a one-meter distance from each other while dining. “Dining out is temporarily prohibited. Keep [a] one-meter distance while queueing (getting in line) and dining. Wash hands before and after dinners,” Luo explains. “[A] disposable tableware policy has also been adopted. We also disinfect tables and chairs after using three times a day. Eat at ease, work at ease.”
Step eight asks for Feelm employees to avoid crowds while commuting. “Walk, ride or drive to work. Try to avoid public transport, if possible,” she says. “If you must, avoid touching anything. Clean phones and keys with wet tissue or medical alcohol often.”
The U.S. Food and Drug Administration has issued warning letters to two companies for illegally selling unapproved products containing cannabidiol (CBD) in ways that violate the Federal Food, Drug and Cosmetic Act. This action is part of the FDA’s efforts to pursue companies that illegally market CBD products with claims that they can treat medical conditions, including opioid addiction or as an alternative to opioids.
“The opioid crisis continues to be a serious problem in the United States, and we will continue to crack down on companies that attempt to benefit from selling products with unfounded treatment claims,” said FDA Principal Deputy Commissioner Amy Abernethy.
“CBD has not been shown to treat opioid addiction. Opioid addiction is a real problem in our country, and those who are addicted need to seek out proper treatment from a health care provider. There are many unanswered questions about the science, safety, effectiveness and quality of unapproved products containing CBD, and we will continue to work to protect the health and safety of American consumers from products that are being marketed in violation of the law.”
The two warning letters were issued to Biota Biosciences of Washington state and Homero Corp DBA Natures CBD Oil Distribution of New Hampshire.
The U.S. FDA has released new guidance for batteries in vapor hardware and bottling improvements for e-liquids.
Battery safety in the vapor industry just got a whole lot better. On Nov. 26, the U.S. Food and Drug Administration (FDA) released an updated premarket tobacco product application (PMTA) guidance for the vapor industry. The new guidance will allow manufacturers of both hardware and e-liquids to provide safer, more effective products to consumers.
“We recognize there are certain modifications manufacturers can make to their tobacco products to address a voluntary industry battery standard and to comply with requirements related to safe packaging of liquid nicotine products, known as flow restrictors,” said Mitch Zeller, director of the FDA’s Center for Tobacco Products. “We encourage these limited safety-related modifications because they are intended to ensure the public is protected from risks such as battery explosions or accidental exposure to toxic levels of nicotine.”
The new guidance, “Compliance Policy for Limited Modifications to Certain Marketed Tobacco Products,” explains the FDA’s compliance policy for making limited safety modifications to vaporizers and e-cigarettes that were on the market as of Aug. 8, 2016, the date all e-cigarettes and other electronic nicotine-delivery system (ENDS) products became subject to the FDA’s tobacco authorities.
The guidance allows for “battery-operated tobacco products modified solely and only to the extent necessary to comply” with the voluntary industry UL 8139 standard for batteries. The FDA worked alongside the U.S. Consumer Product Safety Commission, external stakeholders and UL to develop a voluntary UL 8139 industry standard in order to help manufacturers alleviate potential battery-related risks associated with ENDS products.
A global safety consulting and certification company headquartered in Northbrook, Illinois, USA, UL maintains offices in 46 countries. The 100-year-old company collaborates with a diverse array of stakeholders to establish standards that create level playing fields working to develop new pathways for the latest innovations.
With the new guidance, the FDA is essentially telling vapor companies that the industry should submit batteries used in vapor products to UL for U.S. and Canadian certification, according to Josh Church, chief regulatory and compliance officer with Joyetech Group. Church was a member of the team of scientists and consultants that helped develop the vapor product battery standards and testing guidelines for UL.
The UL 8139 guidelines, titled an “Outline of Investigation for Electrical Systems of Electronic Cigarettes,” evaluates the safety of the electrical, heating, battery and charging systems while also addressing fire safety concerns raised by North American fire officials. The new UL standard has been published with the acceptance of the American National Standard Institute (ANSI) and Canada Standards (CAN). “The acceptance and publication of this standard was a huge landmark for the e-cigarette and vapor industry in general,” said Church.
UL 8139 requirements include determining if lithium cells are operating within safety windows, assessing the battery management system for both normal use and foreseeable misuse, and evaluating compatibility among interconnected systems. It also factors in wide environmental parameters and conditions, tests reasonably expected mechanical stress in use/misuse and requires mechanisms to direct venting away from the inhaler.
UL 8139 standards were written specifically for electronic vapor devices and are part of the larger UL 1642 standards (UL 1642 standards were approved in 2014 by the FDA for use in medical devices) that cover a wide array of lithium-ion products. It’s important to note that neither UL 1642 standards nor UL 8139 standards cover e-liquids or any consumable products used in vaporizer systems. Plus, to qualify for UL 8139 standards, the battery cells must be inside a battery pack, like what you see in a power drill.
This means that the stand-alone standard 18650 e-cigarette battery could never be packaged with “UL” if it’s changeable and not inside a battery pack. However, it can receive a UL “Recognized Component Mark,” or “RU.” This quality mark may be applied to components that are part of a UL listed product but that cannot bear the full UL logo themselves.
The guidance also provides insight into how the FDA wants e-liquids bottled. E-liquid products containing nicotine are now allowed to be modified “solely and only to the extent necessary to comply with the restricted-flow requirements for liquid nicotine containers set out in the Child Nicotine Poisoning Prevention Act of 2015 (CNPPA).” This guidance states that the FDA “does not intend to enforce violations” of the PMTA requirements for manufacturers making these limited modifications.
In order to help lessen the potential risks of accidental exposure to liquid nicotine by children, U.S. Congress passed the CNPPA, which requires liquid nicotine containers to have, among other things, special packaging that makes them difficult for children to open. “The FDA believes e-liquid containers that comply with the flow restrictor requirements will potentially mitigate the risk of children becoming accidentally exposed to toxic levels of nicotine from e-liquids,” a release from the FDA states.
“[This] guidance will provide clarity to manufacturers considering these limited safety-related modifications to their electronic nicotine-delivery system products by outlining our compliance policy for premarket review requirements for such modifications,” said Zeller.
During a recent PMTA seminar/webinar, industry experts say submitting a PMTA will seem overwhelming, but it’s not impossible.
By Timothy S. Donahue
It’s going to be a challenge to get it in on time. It’s going to be even more challenging to get electronic nicotine-delivery system (ENDS) products approved. During a Vapor Voice and TMA joint seminar/webinar held in August, “Finding Direction: Navigating the PMTAs,” in Richmond, Virginia, USA, industry experts discussed the challenges and possible solutions to submitting premarket tobacco product applications (PMTA) to the U.S. Food and Drug Administration (FDA).
Companies will need guidance, according to the panel of eight experts representing the legal, clinical, scientific, nicotine and hardware industries. “You can expect to always be seeking the advice of experts,” a panelist said. “They will provide you options, but ultimately the company will have to make the decisions.” The experts recognized that, currently, there is still a level of uncertainty in the industry on whether any products will be approved. The panel reminded the audience that while the FDA guidance is just a suggestion, the protocols laid out are strongly recommended, and the FDA does not charge a fee for submitting a PMTA.
The process can get overwhelming, they agreed. “However, ultimately, what the FDA wants you to do is to demonstrate that you know your ENDS products,” one panelist suggested. “Your goal is to be able to show that you know and understand what you are selling/producing and that you will always be able to provide further information about your product.” The experts also stated that companies should have a dedicated team or individual in charge of the PMTA process. “You are not going to be successful if you have a part-time person doing this,” one speaker noted.
One of the first issues discussed was the standard set forth by the FDA’s final guidance for vapor and e-cigarette products and the requirement that ENDS must be “appropriate for the protection of public health.” This is under the “Public Health Considerations for ENDS Products” section of the final guidance. Companies must be able to demonstrate that their products are no riskier than the products currently on the market. Companies submitting PMTAs must consider the impact of their product on both users and nonusers, according to the panel. “The most sensitive issue facing vapor products is probably youth use,” a speaker said. “This will be a challenge in light of the recent uptick in youth use.”
Whenever a company is working with the FDA, it is advisable to attempt to look at things from the FDA’s perspective. Many speakers had firsthand experience working with the FDA and agreed this is a viable outlook. “There is the potential for nonusers to use vapor products,” a speaker said. “This can be a difficult thing to overcome …. I think, for the FDA, you must put forward a case that the product is directed toward current smokers and not nonsmokers.”
The statute is extremely broadly worded, an expert noted, adding that it would have been beneficial if the agency issued regulations around the regulatory review process, and even though it said it would, it has not. It’s also important to remember that regulations would be binding while the guidance just offers suggestions, albeit highly recommended suggestions. “It certainly represents what the agency thinks,” a speaker said. “There have been two products to gain FDA approval, but no vapor products have yet to be approved. However, looking at the PMTAs for General snus and IQOS, a heat-not-burn device, will give the vapor industry clues about what the FDA expects in a PMTA submission.”
CONSIDERING THE CONTENT
The second section of the seminar/webinar centered on science. The scientific data a company submits must be solid. “The FDA is not going to be looking for an opinion; the FDA wants data to prove [any statements] about your products,” a speaker explained.
The panel also agreed that studies must be conducted in a valid manner that is generally accepted by the scientific community. The scientific evidence must be based on a unified standard—a methodology that everyone can adopt. It was also recommended to consult the FDA for advice. “If there [are] any questions about whether a study would meet the FDA’s expectations, you need to vet them through a meeting with the FDA. The worst that could happen is if you spend a lot of time and money on a study and discover that it doesn’t meet their expectations for whatever reason,” a speaker said. “You may not get a definitive yes or no, but you will get powerful clues as to whether you are on the right track.”
The experts also said that companies should be submitting data from a comparison product, and that product should be a combustible cigarette. Any applicant would be foolish not to look at comparative products when considering the public health standard, one speaker said. “An applicant needs to look at who they are marketing their product to, who is buying the product and how are they using it,” a speaker explained. “How is your product being used in the real world? What product do you hope to displace? I think those are the most relevant comparators.”
There are several different types of studies that the FDA will expect. The panel said that it is important to remember that while the FDA allows for a single PMTA that covers multiple products, with a combined cover letter and table of contents, the FDA will consider the information included for each product as separate PMTAs. “It is imperative that you clearly identify what content pertains to each distinct product,” according to the FDA. “For example, [the] FDA considers each ENDS product with a differing flavor variant and/or nicotine strength to be a different product.”
The experts agreed that it is critical for companies to be honest and forthright in their PMTA submissions to the FDA. In their submissions, companies will have to explain several aspects of their product from company protocols, management and employee training, consumer complaints, product returns and other standards. For example, companies must explain their packaging and how it will influence the product. There must also be shelf life studies, and all packaging must be child resistant. This data can be used for all products, such as e-liquids, that will require a PMTA submission, according to the speakers.
Some of these studies will be very general. For example, when it comes to required nicotine warning labels, the FDA has laid out what the mandatory warnings are, and companies should have warnings on labeling appropriate for their products. Many of these studies can be bridged, which will help lower costs by using one study across many products, according to the experts. Bridging can also be used within a study across multiple products such as nicotine content (see “All Hands on Deck”). When talking about bridging, “bridging to existing data” and “bridging studies” are two separate concepts, an expert explained after the conference. “These are very different concepts, and both are very important,” he said.
SUBMITTING WITH CONFIDENCE
The FDA is careful about not putting itself into a corner, according to one expert. “The FDA is never going to say, ‘You need to do X, Y and Z and you will get a marketing authorization,’” he said. “They will not be so definitive. They will always give themselves wiggle room.” This is another major reason why a company considering submitting a PMTA should seek professional guidance.
During the final section of the seminar/webinar, the panelists discussed content formatting and any advice they can give companies moving forward. “It helps the relationship with [the] FDA that your PMTA is formatted in a way the FDA is used to,” a speaker said. Another expert said that their company has a guidance document that explains how a document should be put together. “There are a lot of little details …. You want things to be presented consistently so the reader doesn’t get tripped up on locating documents all types of different ways,” the speaker said. “There is a very detailed consideration centered around file naming. It has to be a flat file structure; there can be no folder structure. It is one long list of files from top to bottom.”
Consistency is also key, according to the panelists. Companies should not use different formats in their submissions. “[Often], the data is there but the FDA can’t find it,” a panelist said. “It is always better if you have a simple system.” Giving the FDA the ability to find the necessary information is critical, and the experts agreed that forcing the FDA to “hunt” for information is only going to complicate the process. “Remember this is an electronic submission,” another panelist noted. “My advice is to do whatever makes the most sense for you because the FDA will likely come back the other way …. There has to be a logical and rational [direction in] how you group your data.”
Having a strategy is also important, according to the experts. Companies must have a system in place where they can easily pull data and help the FDA with any questions or concerns about a specific section of a PMTA submission, one panelist noted. “If you don’t have a strategy in place, you are already on a risky path,” she said. “It’s time to put your money down and play your cards.” Another panelist explained that it’s all about minimizing risk and increasing the chances of success. “Take your time, and listen to people you trust,” he said. “There is no set-in-stone rule.”
The PMTA is due May 12, 2020, according to the FDA. The experts agreed that if a company is going to submit a PMTA for an ENDS product, it shouldn’t be a “half-hearted” effort. The entire industry is hampered by this new deadline, including the FDA. “There will be very few companies that will have a ready-to-go-as-intended submission by May,” a speaker explained. “It is the FDA’s view that applications have to be complete on submission. The due date and that statement are at odds with each other.”
Lastly, companies should remember that they will have to continue to submit data to the FDA even after a PMTA is approved. The FDA requires ongoing data submissions—this is not a once complete and approved, it’s finished process. “There will be additional costs, and once a product is approved … you will have an annual requirement to provide the FDA with more information,” a speaker said. “Those [studies] will go on in perpetuity until the FDA rescinds the order … and don’t forget, if a company doesn’t follow through with FDA expectations and requirements, the PMTA can be taken away.” Nothing in the ENDS industry is guaranteed, especially the approval of a PMTA. Like one speaker reminded the audience, “The rules are heavily skewed in favor of the government.”
VAPOR VOICE AND TMA TEAM UP TO SERVE THE INDUSTRY
When the District Court of Maryland recently ordered the U.S. Food and Drug Administration (FDA) to move the premarket tobacco product application (PMTA) deadline forward to May 12, 2020, it sent the vapor industry scrambling.
The new deadline is more than two years ahead of the Aug. 8, 2022 PTMA deadline that was established in 2017 when former FDA Commissioner Scott Gottlieb took over his position—but the burden of compliance is equally daunting.
Vapor companies must carry out expensive and time-consuming studies to demonstrate that their products are “appropriate for the protection of public health”—a considerable hurdle, especially given that the FDA guidance has at times raised more questions than answers.
With only nine months remaining, the pressure to get it right is high. Those who fail to meet the FDA’s strict guidelines will have to remove their products from the market and may even be forced out of business.
To help vapor companies navigate the process, Vapor Voice and TMA organized a one-day workshop in Richmond, Virginia, USA, on Aug. 22. Examining the FDA guidance document chapter by chapter, experts in vapor hardware, clinical testing, data collection and regulation shared their knowledge to help participants make sense of the requirements.
The respective strengths of Vapor Voice and TMA ensured a productive conference. Catering to distributors, manufacturers, retailers and wholesalers, Vapor Voice is a leading source of information on vapor industry regulation, legislation, product development and science.
TMA was established in 1915 to provide unbiased information at a time of policy and industry uncertainty and to act as a convener of forums to address pressing issues. Catering exclusively to the tobacco industry at first, the association has in recent years expanded its scope to cover new nicotine products as well.
TMA’s membership includes manufacturers, retailers, suppliers and service providers.
Mitch Zeller speaking at a TMA annual meeting. Photo: Taco Tuinstra
During this year’s TMA conference, Mitch Zeller, director of the FDA’s CTP, gave his first speech since Scott Gottlieb resigned as FDA commissioner.
It’s business as usual. Mitch Zeller, director of the U.S. Food and Drug Administration’s (FDA) Center for Tobacco Products (CTP), said the agency continues to be “in a position of transition and change” after the abrupt resignation of Scott Gottlieb as the regulatory agency’s commissioner on April 5. “There is some uncertainty,” Zeller said. “But the public health mission of the center does not change.” It was Zeller’s first major public appearance since Gottlieb stepped down.
Speaking to a crowd of more than 130 participants during TMA’s 104th Annual Meeting and Conference, which was held in Falls Church, Virginia, USA, on April 12, Zeller discussed the FDA’s recent reports on a “possible” link between e-cigarettes and seizures. “We don’t yet have causality in these cases, but we have the reports …. We are seeing somewhat of an uptick in reporting … and a seizure is a serious incident,” Zeller said. “In fact, many of the reports we received are incomplete …. We wanted to get the word out to the public but also to those who would be in a position to do a better job of reporting in a more complete way to us.” Zeller then added that within 24 hours of making the announcement, “we have had a whole bunch of additional reports” filed with the agency.
There have been no studies or confirmed reports of any vapor products being the primary cause of a seizure.
Zeller also discussed the FDA’s newly proposed guidance of electronic nicotine-delivery systems (ENDS). The two main focal points surrounding the guidance are flavored e-liquids and pod systems. “The obvious concern, the 800-pound gorilla, is not just Juul, but all Juul-like products—the pod based products, that through some combination of the nicotine levels, the presence of nicotine salts, the flavors and the ease of which these products can be used so discreetly by kids,” Zeller stated. “We remarkably have some kids walking around inaccurately believing there is no nicotine in these products.”
The FDA has stated that if pod systems remain an issue in youth vaping, the products could be removed from the market. Zeller said the FDA has the authority to pull any vapor product from the market because there were no vapor products commercially marketed in the United States as of the predicate date, Feb. 15, 2007.
When asked how the FDA would define a pod-based system, Zeller replied, “It’s a fair question that I’m not going to answer …. The reality is there are no grandfathered ENDS products … there are no ENDS products that have received a marketing authorization through the substantial equivalency [SE] pathway,” said Zeller. “As a technical legal matter, none of these products are lawfully on the market.”
Regarding e-liquid flavors, Zeller said that the FDA looked at all the patterns of use, and, “after taking a tremendous amount of heat from the tobacco control community,” the FDA decided to draw the line at mint, menthol and tobacco flavors. “There’s [mint-, menthol-, tobacco- and non-flavored ENDS], and then there’s everything else, whether it’s grape, strawberry, bubblegum, cotton candy, unicorn puke …. There is a difference because of how popular mint and menthol ENDS products are with adults,” said Zeller. “But because the fruit and candy flavors are so much more popular with kids, not that they aren’t popular with adults, but that is the public health balancing act that we tried to do.”
Vapor products are on the market through an exercise in what the FDA calls “enforcement discretion,” according to Zeller. “That policy has been revisited and revised, which is our right under the law. So, for these other flavored ENDS products (excluding mint, menthol and tobacco) … those products can continue to be sold under our draft guidance,” he said. “But whether it’s in a brick-and-mortar establishment or online, there has to be some form of heightened age restriction that can prevent kids from getting access.”
The FDA has undertaken more than 1 million compliance checks since 2010, according to Zeller. He added that the 17 companies that were recently given warning letters concerning e-liquids that could be seen as being marketed toward youth had all “completely reformed.” Issues persist, however, Zeller said. “We had to send these warning letters [recently to a company] because of the resemblance between [their] e-liquid and a heavy duty, codeine-laced prescription drug, and the concern here was ingestion … because of the resemblance … you see the deliberate resemblance in the labeling,” he said.
During the question and answer session of his speech, Zeller was often forced to say, “I can’t comment on that right now.” When one attendee asked about the recent postponement of harmful and potentially harmful constituent testing, Zeller said, “I think I’m going to need to continue to disappoint you. We are working on it, and I can’t say anything more than that, other than we are now on record … saying that whatever the reporting requirements will be, it won’t be until after there is a final draft guidance …. Let’s just say [that] when it comes to clearance, we don’t control our own destiny … when it comes to getting things through the process.”
Bonnie Herzog, managing director of Wells Fargo Securities, wrote in an email that Zeller was understandably “careful to refrain from making any comments” regarding most of the policies the FDA has proposed in its recent rules. “However, to us, the important nuance was what director Zeller didn’t say. Namely, he refrained from repeating former commissioner Gottlieb’s strong threats to take more decisive action on youth e-cig access (e.g., removal of e-vapor pods from the market),” said Herzog. “This nuance suggests a distancing from Gottlieb’s more severe approach in anticipation, we think, of a potentially different tone from the new acting FDA commissioner …. In sum, the conference illuminated the complexity of the issues before the FDA …. It is clearly a delicate balancing act that remains elusive.”
What does Gottlieb’s departure mean for the vapor industry? The verdict isn’t in yet, but it’s looking like a blip on the map.
By Maria Verven
It was an announcement that took many by surprise—and may have even brought a glimmer of hope to some. When Scott Gottlieb announced he would be stepping down as commissioner of the U.S. Food and Drug Administration (FDA), it marked the end of a long and heated battle between the commissioner and the vapor community.
While it remains to be seen how this battle will unfold, one thing is for certain: Gottlieb’s departure won’t signal the end. The fight will rage on.
PARTING SHOTS: CHANGING THE COMPLIANCE DATE
Before his departure, Gottlieb made certain he left his imprint on key regulations going forward.
In mid-March, the FDA distributed its “modifications to compliance policy for certain deemed tobacco products.” Citing the “public health threat” from the “epidemic rise in youth use” of ENDS—electronic nicotine-delivery systems such as vaporizers or e-cigarettes—the modifications included changes to both compliance policies and deadlines for e-cigarettes.
The modifications were necessary, the FDA argued, because ENDS pose a greater risk for minors. Thus, the compliance date for submitting premarket tobacco product applications (PMTAs) for ENDS—except for tobacco-, mint- and menthol-flavored products—was moved up one year to Aug. 8, 2021.
After this date, the FDA says it intends to “prioritize enforcement” against manufacturers of flavored ENDS products (except tobacco, mint or menthol products) that have not submitted a PMTA, regardless of any steps the manufacturer took to prevent minor access and appeal.
“By promoting submission of premarket [tobacco product] applications for flavored ENDS products by Aug. 8, 2021, [the] FDA is recalibrating its balancing of public health considerations in light of the public health threats (i.e., underage use).
“This new policy reflects [the] FDA’s balancing of concerns regarding the appeal of certain flavored ENDS products to youth, the potential public health benefit of noncombustible options in helping transition adult smokers completely off of combustible tobacco products, and the uncertainty created by extended availability of these new tobacco products without scientific review and evaluation under the public health standard,” the document states.
THE QUEST TO CURB TEENAGE VAPING
The FDA’s campaign against underage use is being fought on multiple fields. The Smoke-Free Alternatives Trade Association (SFATA) actively supports age restrictions on vapor products. However, it also vehemently opposes what it considers to be misguided efforts that end up prohibiting sales to adults.
“We will actively oppose bills that seek to prevent legal access to our products for adults by raising the legal age to purchase higher than that at which Americans can also enter into legally binding contracts, build financial debt, get married, be drafted into military service or be held accountable for their actions as adults in the courts,” the SFATA’s Age to Vape policy states.
“I am confident that these efforts by the FDA to curb underage vaping will continue, and we support most of them,” says Mark Anton, SFATA executive director. “But our major battle will be fighting off flavor bans and the false notion that flavors are driving youth use. Banning flavors for adults who rely on them would be a bad policy for harm reduction and will devastate the whole industry.”
Last May, the FDA issued 17 warning letters to manufacturers, distributors and retailers for selling e-liquids with labeling and/or advertising that resembled those found on kid-friendly foods such as juice boxes, candy and cookies. Last fall, after conducting nationwide undercover investigations of brick-and-mortar stores and online retailers, the agency issued more than 1,300 warning letters and civil money penalty complaints to retailers that illegally sold ENDS products to minors, as well as 12 warning letters to online retailers that sold e-liquids whose labeling and/or advertising resembled that found on kid-friendly food products.
The FDA also issued letters to five ENDS manufacturers, including Juul Labs, requesting that they submit a plan describing how they would prevent and prohibit use of their products by minors.
Manufacturers described a number of safeguards that would restrict minors’ access to their products, including using independent, third-party age- and identity-verification services using public records or other third-party data sources. Other strategies included limiting the quantity of ENDS products customers may purchase within a certain period of time.
However, proposals that would restrict the type of stores in which ENDS products can be sold fail to meet any fairness test, say industry supporters.
“So long as cigarettes remain legally available to purchase, it is preposterous to force adult smokers to seek out a tobacco or vape shop to buy a product that can help them quit,” says Gregory Conley, president of the American Vaping Association. “Plus, if this policy fails to drastically reduce youth use, the calls for flavor prohibition will only grow stronger.”
THE NUMBERS DON’T ADD UP
At the very heart of the battle is the concern that teenagers are using e-cigarettes. The FDA’s new compliance policy cites the 2018 National Youth Tobacco Survey (NYTS) and a supposed significant increase in youth use of ENDS products, particularly in the past year: “For example, data from the NYTS [shows] that, between 2017 and 2018, current e-cigarette use among high school students increased 78 percent (11.7 percent to 20.8 percent, p<0.05).”
The problem here is that the data doesn’t show that 20.8 percent of high school students are current e-cigarette users. The data shows that 20.8 percent of kids have ever used an e-cigarette. In fact, only 3.7 percent had used an e-cigarette for 20 or more days. And of the students that were polled, 8.9 percent of them were 18 years or older and therefore could legally purchase and use an e-cigarette.
The U.S. Centers for Disease Control and Prevention (CDC) has also sounded what some might construe to be a false alarm on the “epidemic” of teen vaping, citing the same NYTS data. At least the CDC is clear on how it defines “current” use. Buried in their findings is this statement: “Current e-cigarette use was defined as a response greater than [zero] days to the question, ‘During the past 30 days, on how many days did you use e-cigarettes?’”
So with these criteria, a teenager who takes one puff of an e-cigarette at a party one night would be classified as a “current” user.
Another study supposedly focused specifically on youth use of the brand Juul, which currently has the largest share of the ENDS market. Yet the study surveyed participants in a surprisingly wide age range—ages 15 to 34. Still, only 6 percent reported ever using the Juul, and only 3.3 percent reported use in the past 30 days (i.e., supposed “current” use).
“Since the massive media blitz on youth use—which is still 80 percent infrequent experimentation, Gottlieb changed his tune dramatically to the point where he was willing to sacrifice the lives of adult smokers to prevent youth experimentation,” Anton said. “The false narrative that flavors are the driving force for youth experimentation shows a disconnect with real-world factors and is having unintended consequences.”
MISSING THE WHOLE POINT
While Gottlieb rightly sounded the alarm on the dangers of traditional cigarettes, his near obsession with underage vaping obliterates the benefits of e-cigarettes to adult smokers and where they fall on the risk continuum.
“During Dr. Gottlieb’s tenure, there has been no shortage of issues on which to criticize him,” Conley said. “As commissioner, he failed to fulfill several goals that he laid during his July 2017 speech on nicotine regulation, including fostering an accurate understanding of nicotine’s health effects and bringing more transparency and efficiency to the FDA’s PMTA system.”
Fortunately, recent studies, such as the one published in January 2019 in the New England Journal of Medicine, continue to provide legitimate scientific evidence of the product’s benefits.
This study found that e-cigarettes were nearly twice as effective as traditional nicotine-replacement therapy products, such as patches and gums.
“Health professionals have been reluctant to recommend their use because of the lack of clear evidence from randomized controlled trials. This is now likely to change,” said Peter Hajek, the study’s lead author and a clinical psychology professor at Queen Mary University of London, which coordinated the clinical trials through public stop-smoking clinics.
“The hazard to health arising from long-term vapor inhalation from the e-cigarettes available today is unlikely to exceed 5 percent of the harm from smoking tobacco,” states the most recent policy by the U.K.’s Royal College of Physicians. “In the interests of public health, it is important to promote the use of e-cigarettes as widely as possible as a substitute for smoking in the U.K.”
Yet the FDA still refuses to make factual risk communications, according to Anton. “And when we asked, they told us that truthful statements on risk are too complicated for the public to understand or that they are ‘youth permissive,’” he says. “They seem to think that if we say that the Royal Society for Public Health says the effects of nicotine on human health are the same as caffeine, it will encourage more youth to vape. So they refuse to have honest communications with the public. In addition, anti-tobacco groups have pushed the narrative that vaping is as bad for you as smoking, or that we are using the same tactics as Big Tobacco to attract youth use. “We are certain these groups will exert extreme pressure on the administration to appoint someone who is anti-vape.”
“Right now, we have more questions than answers on what the future of the FDA will look like without Dr. Scott Gottlieb at the helm,” Conley says. “We are hoping that key Republican lawmakers like Senators Richard Burr and Thom Tillis will personally call President Trump and inform him of the importance of appointing an FDA commissioner that won’t so easily kowtow to anti-vaping activists.”
“It will take a culture change at the FDA to change their approach—and more than just a change at the helm,” says Anton.
Maria Verven
The original “Vaping Vamp,” Maria Verven owns Verve Communications, a PR and marketing firm specializing in the vapor industry.
In the struggle against underage vaping, restricting access to flavored e-cigarettes should be the last resort.
By Chris Howard
Put yourself in the shoes of a smoker who is trying to quit. You’ve tried various quit-smoking products, such as nicotine patches, gums and prescription medications. None have worked. Your friend tells you how vaping with flavors works, so you decide to give e-cigarettes a try.
You stop at a convenience store, seeking a vapor product that meets your needs. Surprisingly, the flavors that seemed so appealing are nowhere to be found. You never liked menthol, and tobacco reminds you of the cigarettes you hope to avoid. Like many smokers, you’re anxious about leaving cigarettes behind and your ability to quit. This latest obstacle makes quitting seem impossible (again). What happens next? You buy another pack of cigarettes and promise to try again another day.
The reality is that smokers don’t get to count on “another day.” Each cigarette increases the smoker’s risk of developing a smoking-related illness that will inevitably lead to a lower quality of life or worse—premature death.
The vapor industry shares the U.S. Food and Drug Administration’s (FDA) goal of ensuring e-cigarettes are used only by adults. That said, the FDA should consider a variety of more reasonable options to curb youth access without jeopardizing e-cigarettes’ harm reduction potential.
Codify marketing standards: Many adult-oriented industries employ voluntary codes of conduct to govern marketing practices. The FDA should seek consensus standards prohibiting marketing practices directed at youth. The Vapor Technology Association (VTA), which advances the interests of 800-plus manufacturers, wholesalers, small-business owners and entrepreneurs of the vapor industry, has already enacted such standards (see https://bit.ly/2Ozbfyn), which are mandatory for membership. Most, if not all, vapor companies would likely embrace objective, FDA-accepted parameters as reasonable ways to reduce youth e-cigarette access.
Revamp enforcement scheme: Undoubtedly, the FDA is resource-constrained and overwhelmed by age-verification requirements for vapor products. That said, even if the FDA could act on every violation, the current scheme allows a retailer to sell tobacco products to a minor five times within a 36-month period before the FDA can stop them from selling tobacco for 30 days. Revamping the current enforcement policy to provide for stricter and more consistent penalties would enable the FDA to use agency resources more effectively and deter bad actors from breaking the law.
Enhance restrictions for online purchases: Some online retailers are already implementing two-factor authentication and other measures to curb youth access to vapor products—measures rightly considered by the FDA. Such online purchase restrictions should continue to be developed, strengthened and exercised in order to eliminate youth access to vapor products.
Work with schools: Nearly every anecdotal news report involves a teenage student’s exposure to e-cigarettes. Given that schools are also seeking tools to combat this issue, the FDA should work more closely with school districts to provide resources and training that helps teachers identify the signs of youth e-cigarette use and how to stop it. The FDA’s current “The Real Cost” campaign is the right approach to assist teachers and educate youth, but unfortunately the messaging is riddled with inaccuracies about health effects that could deter smokers of combustible cigarettes from switching to vapor products.
Analyze the impact of higher minimum purchase age: Some states have increased the minimum purchase age for e-cigarettes to 21. The FDA should analyze purchase and use patterns in these states to assess any reduction in straw sales and youth usage rates resulting from increased purchase age requirements.
The decisions the FDA makes affect millions of adults who rely on vapor products for harm reduction. Restricting the availability of flavors is a radical measure that places unnecessary obstacles for adults obtaining products of their choice. In many ways, the proposed restrictions may actually help combustible cigarettes.
For these reasons, the FDA’s proposed flavor ban should be a last resort.
Chris Howard
Chris Howard is vice president, general counsel and chief compliance officer at E-Alternative Solutions.
Nineteen advocacy organizations ask the U.S. President to halt the FDA’s attack on vapor products.
By Timothy S. Donahue
It was necessary. On Feb. 4, a letter signed by representatives from 19 advocacy groups was sent to U.S. President Donald Trump urging him to immediately end the U.S. Food and Drug Administration’s (FDA) “aggressive regulatory assault” on the vapor industry.
“From inaction on pending product approvals to threatening letters sent to American manufacturers and promises to begin new rulemaking that would make illegal certain consumer products, this FDA is currently pursuing several policies that are more extreme than those contemplated by the Obama administration,” the letter reads. “FDA Commissioner Scott Gottlieb’s effort to curb the $6.6 billion electronic cigarette industry and an even larger reduced-risk tobacco alternatives market is inconsistent with your clearly articulated deregulatory objectives and will destroy jobs, limit consumer freedoms and harm public health.”
The signatories were: Grover Norquist, president of Americans for Tax Reform (ATR); Lisa Nelson, CEO of ALEC Action; Norm Singleton, president of the Campaign for Liberty; Tom Schatz, president of Citizens Against Government Waste; Michelle Minton, senior fellow for the Competitive Enterprise Institute; Jeff Stier, senior fellow for the Consumer Choice Center; Kenneth T. Cuccinelli II, director of Regulatory Action Center for FreedomWorks and former Attorney General of Virginia; Henry I. Miller, former director of the Office of Biotechnology at the FDA; Naomi Lopez Bauman, director of Healthcare Policy at the Goldwater Institute; Mario H. Lopez, president of the Hispanic Leadership Fund; Julie Gunlock, director of the Center for Progress and Innovation for the Independent Women’s Forum; Bob McClure, president and CEO of The James Madison Institute; Seton Motley, president of Less Government; Pete Sepp, president of the National Taxpayers Union; Douglas Kellogg, director of Ohioans for Tax Reform; Carrie L. Wade, director of Harm Reduction Policy at the R Street Institute; Paul Gessing, president of the Rio Grande Foundation; and Tim Andrews, executive director for the Taxpayers Protection Alliance.
The letter explains that vapor products do not contain tobacco and that they deliver nicotine without the combustion or tar that is found in traditional cigarettes. There are also numerous studies that have found that vapor products are at least 95 percent less harmful than combustible cigarettes. The letter also states that Gottlieb, the FDA Center for Tobacco Products’ director, Mitch Zeller, and Surgeon General Jerome Adams have acknowledged the harm reduction potential of e-cigarettes.
“Unfortunately, a spike in the use of these products by teens has resulted in regulatory panic and significant government overreach,” the letter reads. “Commissioner Gottlieb has already pressured major manufacturers of e-cigarettes to remove many products from convenience store shelves, suggested that more than 100,000 retailers limit adult access to these products and threatened to use agency power to remove thousands of legal products from the market. We do not write you today urging your administration to ignore the concerns about the use of e-cigarettes by teens. We do, however, urge your administration to subject the FDA’s response and actions to much closer scrutiny and examine it within the context of your broader deregulatory and pro-jobs agenda.”
When President Trump signed executive order 13771, “Reducing Regulation and Controlling Regulatory Costs,” he directed departments and agencies to not only eliminate at least two regulations for every new one created but to sensibly manage costs. The deregulatory efforts led to over $33 billion in savings through October 2018, according to the letter. Nearly every department and agency identified harmful regulations and worked to untangle and repeal them. One glaring exception has been the FDA.
“It’s important that we hold the president accountable for the promises he made in the 2016 campaign and initial days of his administration,” said Paul Blair, director of strategic initiatives for ATR. “Regardless of one’s politics, it’s clear that across every department and agency, the deregulatory agenda is being fully implemented. That’s just not happening at the FDA, and we want the president to know that conservatives are fed up.”
Blair goes on to say that vapers are passionate consumers, and, more importantly, they represent millions of voters, adding that they believe they’ve made a personal decision to improve their health. “There is a broad coalition in support of our efforts here; it’s not just [ATR’s] discontent at the FDA,” he says. “I want the president to understand that even though ATR has been out in front of this issue for years, we’re not alone in recognizing the importance of getting regulations for the tobacco and vapor industry right.”
It is likely that the impact of the FDA’s proposed, pending, and possible new guidance and rules for vapor products will amount to billions of dollars in lost economic activity and costs, according to the letter. Blair says it’s inexcusable. “At this point, a Hillary Clinton presidency would have been no different for the industry than Trump’s, all thanks to Scott Gottlieb’s misguided crusade.”
STUDY TIME
They keep coming. Another positive study concerning vaping has been completed. On. Jan. 30, the New England Journal of Medicine published a predominantly U.K.-based study that finds that “e-cigarettes [are] more effective for smoking cessation than nicotine-replacement therapy when both products [are] accompanied by behavioral support.”
For the study, scientists randomly assigned adults attending the U.K. National Health Service stop-smoking services to either nicotine-replacement therapy (NRT) products of their choice, including product combinations, or an electronic nicotine-delivery system (ENDS) starter pack, with a recommendation to buy their own flavors and strengths of e-liquids. The treatment plans also included weekly behavioral support for a minimum of four weeks.
The researchers wanted test subjects to have sustained abstinence for one year, which was validated biochemically during the patient’s final visit. A total of 886 participants were involved. The one-year abstinence rate was 18 percent in the ENDS group, according to the study, as compared with 9.9 percent in the NRT group.
Among subjects with one-year abstinence, those in the ENDS group were more likely than those in the NRT group to use their assigned product at 52 weeks (80 percent [63 of 79 participants] vs. 9 percent [four of 44 participants]), according to the study. Overall, throat or mouth irritation was reported more frequently in the e-cigarette group (65.3 percent vs. 51.2 percent in the NRT group) and nausea more frequently in the nicotine-replacement group (37.9 percent vs. 31.3 percent in the e-cigarette group).
The researchers hail from Queen Mary University of London; King’s College London; London South Bank University, London; the University of York, York; Leicester City Council, Leicester; and the Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA. –T.S.D.